|
||||
|
|
ПРИЛОЖЕНИЕ ОБМЕН МОНОАМИНОВ И ПСИХИЧЕСКИЕ НАРУШЕНИЯ Так как действие ряда лекарств, о влиянии которых на психику говорилось в первой части этой книги, а также действие ряда психотомиметичеоких средств пытаются объяснить как результат их влияния на обмен серотонина и адреналина, мы приводим в виде приложения краткие сведения об обмене серотонина и адреналина, их взаимодействии с лекарствами и роль при психических заболеваниях. Глава 1 ОБМЕН СЕРОТОНИНА И ПСИХИЧЕСКИЕ НАРУШЕНИЯ В 1948 году Раппорт, Грин и Пейдж выделили из крови вещество, обладающее сосудосуживающим действием, и назвали его серотонином. Примерно в то же время Эрспамер и Азеро обнаружили в слизистой желудка и кишечника, коже и слюнных железах низших животных (лягушек, саламандр) вещество, которому дали название энтерамин. Дальнейшие исследования показали, что речь идет об одном и том же веществе — 5-гидрокситриптамине. В дальнейшем серотонин был обнаружен в различных органах и тканях человека и животных. Количество работ, посвященных изучению этого вещества и роли, которую оно играет в организме, в настоящее время чрезвычайно велико. Мы не останавливаемся на вопросе о роли серотонина в регуляции тонуса сосудов, функции пищеварительной системы, аллергии и т. д. и ограничиваемся лишь рассмотрением данных о той роли, которую играет серотонин в функционировании центральной нервной системы, в первую очередь, головного мозга, и, следовательно, в нарушениях психической деятельности человека. У ряда экспериментальных животных небольшие концентрации серотонина были обнаружены в головном мозгу. При этом оказалось, что в различных отделах мозга серотонин распределяется неравномерно. Он концентрируется почти исключительно в сером веществе головного мозга, причем в подкорковых областях концентрация его выше, чем в коре головного мозга и в мозжечке. Наивысшая концентрация серотонина обнаружена в гипоталамической области и в ретикулярной формации ствола головного мозга. Затем следуют в убывающем порядке обонятельный мозг (в первую очередь, пириформная доля и миндалевидные ядра) и мезэнцефалон. Таким образом, распределение серотонина в ЦНС сходно с распределением в ней норадреналина и адреналина. Примерно таким же образом распределяются в ЦНС и основные ферменты, участвующие в образовании и разрушении серотонина — 5-гидрокситриптофандекарбоксилаза и моноаминоксидаза (МАО), хотя полного совпадения не обнаружено. Так, 5-гидрокситриптофандекарбоксилаза лишь в небольших количествах содержится в пириформной доле и в миндалевидных ядрах, богатых серотонином, напротив, в симпатических ганглиях содержится большое количество декарбоксилазы, а концентрация серотонина низка. МАО распределена по всему серому веществу головного мозга, но максимальная концентрация ее — в гипоталамусе, наиболее богатом серотонином. Что касается распределения серотонина внутри нервной клетки, то данные по этому вопросу противоречивы. МАО локализуется преимущественно в митохондриях цитоплазмы, где по некоторым данным содержится и большая часть — 2/3 — серотонина, декарбоксилаза содержится в растворимой фракции цитоплазмы нервной клетки. Существует предположение, что 5-гидрокситриптофандекарбоксилаза идентична диоксифенилаланиндекарбоксилазе, участвующей в образовании катехоламинов. Химическая формула, основные пути образования и распада серотонина, в частности, в ЦНС, в настоящее время известны, хотя и не полностью. Источником образования серотонина является триптофан, часть которого под действие триптофангидроксилазы превращается в 5-гидрокситриптофан (5-ГТФ), из которого под влиянием 5-ГТФ-декарбоксилазы образуется серотонин (5-гидрокситриптамин). Пиридоксал-фосфат является коферментом декарбоксилазы, поэтому при недостатке витамина B6 образование серотонина из 5-ГТФ может нарушаться. Так, Вайсбах, Богдански и сотр. показали, что у цыплят при недостатке витамина B6 в организме содержится гораздо меньше серотонина, чем у контрольной группы. Гидроксилазы, способные превращать триптофан в 5-ГТФ, в мозгу не обнаружены, но мозг содержит декарбоксилазы, способные превращать 5-ГТФ в серотонин. Поэтому считают, что 5-ГТФ образуется вне ЦНС, а затем, проникая в ЦНС, декарбоксилируется и превращается в серотонин. 5-ГТФ легко проникает через гемато-энцефалический барьер — это доказано многочисленными экспериментами, в которых как прием внутрь, так и парентеральное введение 5-ГТФ резко увеличивало содержание в головном мозгу серотонина (что, кстати подтверждает и то обстоятельство, что 5-ГТФ предшественник серотонина). Принято считать, что серотонин, в отличие от 5-ГТФ, не проходит через гемато-энцефалический барьер и образуется из 5-ГТФ непосредственно в головном мозгу. Действительно, в части экспериментов на различных животных эффект серотонина удавалось получить только при его внутрижелудочковом введении, тогда как парентеральное введение серотонина не оказывало влияния на ЦНС. Однако, непроницаемость гемато-энцефалического барьера для серотонина относительна. В некоторых экспериментах, как например, в опытах на кроликах, проведенных Коста и Эприсоном, опытах на крысах Кэрки и Паасонена и др. отмечено увеличение содержания серотонина в головном мозгу после внутривенного его введения. Основным ферментом, расщепляющим серотонин, является МАО под влиянием которой серотонин превращается в 5-гидроксииндолуксусную кислоту (5-ОИУК, по-видимому, проходя через стадию образования 5-гидроксииндолуксусного альдегида), которая выделяется из организма, главным образом, с мочой. Схематически обмен серотонина может быть представлен следующим образом: 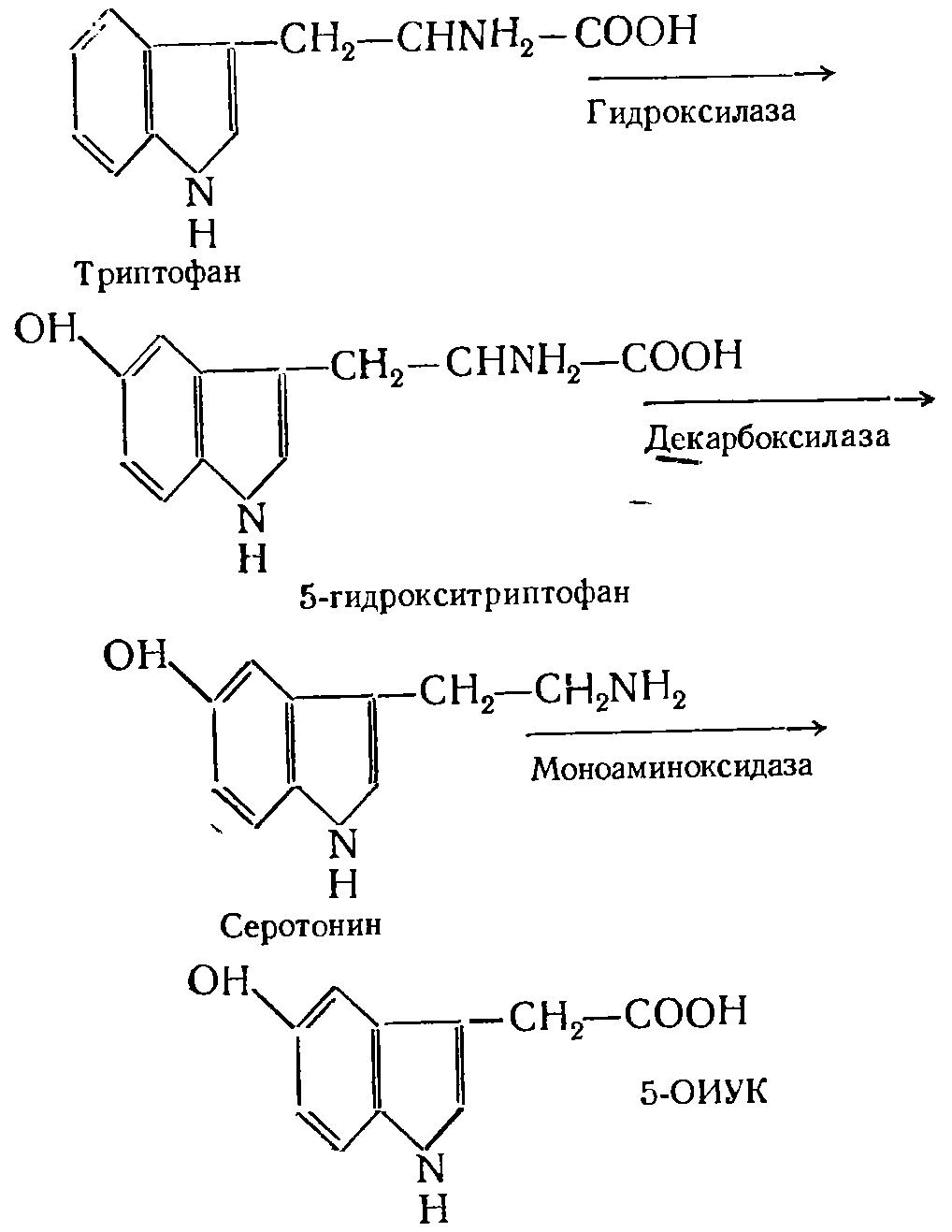 В действительности обмен серотонина сложнее, чем на представленной схеме. Наряду с дезаминированием может происходить: 1) образование парных соединений с глюкуроновой и серной кислотой, 2) образование 5-О-триптамина, 3) превращение 5-гидроксииндолуксусного альдегида не в 5-ОИУК, а в коричневый пигмент, 4) N-ацетилирование, 5) N-метилирование (показанное главным образом у беспозвоночных). Наконец, часть серотонина и 5-ГТФ выделяется с мочой в неизменном виде. Выделяющаяся с мочой 5-ОИУК составляет всего 20—40% метаболизировавшегося серотонина. Вместе с тем, образование 5-ОИУК может, очевидно, происходить и за счет действия не только МАО, но и других энзимов. Так, Вайсбах, Ловенберг и сотр. в опытах на мышах показали, что при полном блокировании МАО образование 5-ОИУК продолжается (хотя количество ее уменьшается) и одновременно возрастает образование серотонин-О-глюкуронидов. Таким образом, но концентрации и общему количеству содержащейся в моче 5-ОИУК нельзя составить полного представления об интенсивности обмена серотонина. Правда, серотонин в ЦНС почти целиком расщепляется за счет МАО, однако серотонин в ЦНС составляет лишь около 1 % по отношению к серотонину, содержащемуся во всем организме, и лишь около 5% всей 5-ОИУК образуется за счет метаболизма серотонина в головном мозгу. Так как период полураспада серотонина в головном мозгу значительно короче, чем в других органах (считают, что он равен всего нескольким минутам), некоторые исследователи предполагают, что изменения выделения 5-ОИУК с мочой могут отражать изменения серотонина в головном мозгу. Однако это относится только к случаям значительного усиления обмена серотонина в ЦНС, которое может вести к увеличенному выделению 5-ОИУК с мочой. Если же распад серотонина в головном мозгу замедлен или даже полностью блокирован, то падение выделения 5-ОИУК будет крайне незначительным (если метаболизм серотонина в других органах не нарушен) и не превысит пределов ошибки, допускаемой точностью применяемых для ее определения методик. Данные, касающиеся обмена серотонина в ЦНС, получены на животных, проведение таких исследований на людях практически невозможно — за исключением исследований на трупах и на участках головного мозга, удаленных во время операции. Поэтому подавляющее большинство исследований на людях проведено с помощью определения содержания серотонина в крови и 5-ОИУК в моче. Немногочисленные исследования спинномозговой жидкости показали, что концентрация в ней серотонина значительно ниже, чем в крови, и в ряде случаев настолько низка, что не улавливается современными методами исследования. Это отчасти подтверждает непроницаемость или неполную проницаемость гемато-энцефалического барьера для серотонина и у человека. Большое количество экспериментов проведено на животных путем непосредственного введения серотонина в желудочки мозга, наложения его на поверхность участков головного мозга или парентерального введения предшественника серотонина — 5-ГТФ. Эти опыты показали, что серотонин оказывает центральное действие и, следовательно, является веществом активно участвующим в деятельности головного мозга. Введением больших доз серотонина удавалось вызвать кататонические симптомы в виде обездвиженности, отсутствия реакции на раздражитель, длительного сохранения приданных поз. Такие состояния были получены у рыб, лягушек, мышей, цыплят; при непосредственном введении в желудочки мозга, — у кошек и собак, хотя и не во всех экспериментах. В частности, Бонамини и Гарелло отметили, что при повторных введениях серотонина в желудочки мозга никогда не развивается хроническая кататония. В ряде других наблюдений двигательные изменения были не столь резко выражены и эффект серотонина выражался лишь в уменьшении моторной активности, состояниях прострации. Центральное тормозящее действие серотонина проявляется также и в потенцировании действия успокаивающего средства — мепробамата (отечественный мепротан); в увеличении продолжительности сна, вызываемого различными барбитуратами или хлоргидратом у мышей, крыс, и кроликов; в подавлении условного рефлекса бегства, который у крыс и мышей под влиянием серотонина вырабатывается с значительно большим трудом и становится непостоянным, тогда как безусловный рефлекс не меняется; в уменьшении реакции пробуждения и первичного ответа на ЭЭГ. Аутоинъекции быстро прекращаются из-за наступления сна, на аутостимуляцию серотонин не влияет[5]. Однако, такой характер действия серотонина наблюдается не всегда и может изменяться с увеличением дозы. Так, после введения больших количеств 5-ГТФ у животных развиваются состояния возбуждения с утратой или ослаблением реакции на внешние раздражители, дезориентировка, состояния страха или ярости, тремор, тахикардия, мидриаз, пилоэрекция, слезо- и слюнотечение, одышка, артериальная гипертензия и одновременно появление реакции пробуждения на ЭЭГ. У наркотизированных животных эта реакция отсутствует. Отсутствие и в этих случаях корковых и подкорковых реакций на раздражение ретикулярной формации позволяет предположить, что она и в этих случаях остается заторможенной. Таким образом, малые дозы серотонина оказывают депрессорное действие на ЦНС, а большие — стимулирующее. Это положение не является бесспорным — в отдельных экспериментах, как например в опытах Ферро-Милоне и Гомирато состояния возбуждения и десинхронизация ЭЭГ становились менее выраженными с увеличением дозы серотонина. Поскольку в большинстве опытов, в которых у животных наблюдалось состояние возбуждения, вводили не серотонин, а 5-ГТФ, нельзя исключить предположение о связи возбуждения не с накоплением серотонина, а с образованием из части введенного 5-ГТФ триптамина. Данные о противосудорожном действии серотонина противоречивы. С одной стороны, отмечено защитное действие серотонина по отношению к судорогам, вызываемым электрошоком, звуком, кислородом, вводимым под давлением кардиазолом, пикротоксином. Снижение судорожного порога под влиянием резерпина и нитомана (тетрабеназина) некоторые авторы связывают с вызываемым этими препаратами снижением содержания в ЦНС серотонина. С другой стороны, отмечены судорожное действие больших доз серотонина, как например, в эксперименте Вада, которая наблюдала у кошек и обезьян усиление судорожной активности на ЭЭГ после введения серотонина и пришла к выводу, что серотонин повышает судорожную активность в головном мозгу, а эпилептогенный очаг повышенно чувствителен к серотонину. В ряду экспериментов — хотя опять-таки не во всех — обнаружено повышение концентрации серотонина в головном мозгу после судорожного припадка. Эти данные указывают на участие серотонина в деятельности ЦНС, но не доказывают и не опровергают предположение о противосудорожном эффекте серотонина. О центральном действии серотонина свидетельствуют также опыты, в которых обнаружено его влияние на активность холинэстеразы мозга (хотя по некоторым данным серотонин усиливает, а по другим — тормозит эту активность), задержка воды тканью коры головного мозга (кроликов) под влиянием серотонина в опытах in vitro, облегчение проведения возбуждения через ганглии, антагонизм по отношению к серотонину ряда веществ, оказывающих влияние на ЦНС (эти данные рассматриваются ниже), изменения поведения животных под влиянием некоторых антагонистов серотонина, изменения ЭЭГ при местном наложении серотонина на участки коры головного мозга. В серии опытов, проведенных на кошках, Маррацци, Харт и сотр. нашли, что серотонин вызывает торможение в синапсах, превосходя большинство веществ, оказывающих такое же действие. В частности, тормозящее действие серотонина оказалось в 20 раз сильнее действия адреналина и в 2000 раз сильнее действия адренохрома. Это тормозящее действие серотонина в головном мозгу отчетливее, чем в низших отделах ЦНС. В этих опытах исследовалась транскаллозальная (через мозолистое тело) передача возбуждения с раздражаемого участка коры головного мозга на гомологичный участок другого полушария. Торможение в синапсах по Маррацци и Харту играет решающую роль в генезе психических заболеваний. Важное значение придается другой группой авторов сокращениям клеток микроглии, вызываемым серотонином. Эти сокращения наблюдали в культуре мозговой ткани Вулли и Шоу (их данные подтверждают и другие исследователи), которые считают, что, сокращая клетки микроглии, серотонин участвует в перемещении жидкости и питательных веществ в головном мозгу, обеспечивая питание нервной клетки, которое нарушается при разрушении серотонина. Если все накопленные в настоящее время данные позволяют считать серотонин веществом, играющим роль в жизнедеятельности головного мозга, то характер его действия все еще остается неясным. Отмечающаяся нами выше противоречивость экспериментальных данных отчасти зависит от разной чувствительности к серотонину разных видов животных, отчасти от разницы в дозах, существенную роль играет и то обстоятельство, что часто определяется лишь суммарная концентрация серотонина в головном мозгу, а не взаимоотношение отдельных его фракций. Дача внутрь или парентеральное введение серотонина психически здоровым людям (в том числе, страдающим гипертонической болезнью) обычно вызывает только соматические изменения преимущественно сосудистого характера: быстрый подъем систолического давления (у больных гипертонической болезнью — его снижение), тахикардию, чувство жара или холода в конечностях, головокружение, головную боль, чувство сдавления в области сердца, снижение мышечного тонуса. Психическое состояние и характер ЭЭГ после приема серотонина не изменялись. Менее однородны данные, полученные после приема серотонина у психически больных. Часть исследователей не обнаружила никаких изменений в психическом состоянии больных, преимущественно шизофренией. В то же время при различных психических заболеваниях, в том числе, при шизофрении, эпилепсии, депрессивных состояниях, психоневрозах ряд исследователей отметил изменения ЭЭГ (напр., некоторое замедление и большая регулярность ?-ритма у больных шизофренией и усиление патологических изменений на ЭЭГ у больных эпилепсией по наблюдениям Полони), а также различные изменения их психического состояния. Эти последние обычно были кратковременными и не оказывали сколько-нибудь заметного влияния на течение основного заболевания. Так, описаны общее седативное действие, улучшение внимания и качества выполнения психологических тестов, обострение тревоги, бессонница, сглаживание или исчезновение апатии и астении, смена депрессивного состояния маниакальным. В отдельных случаях под влиянием серотонина наступало общее улучшение психического состояния и даже выздоровление. Так как считают, что серотонин не проникает через гемато-энцефалический барьер, отсутствие эффекта при его приеме внутрь или при внутривенном введении может быть обусловлено тем, что серотонин в этих случаях не достигает ЦНС. Поэтому были проведены пробы с эндолюмбальным введением серотонина или чаще — парентеральным введением или приемом внутрь предшественника серотонина — 5-ГТФ. Эти попытки также не дали отчетливых результатов. Нам известны всего 2 работы, в которых приведены результаты эндолюмбального введения серотонина; Платаниа и Катанцаро не отметили никаких психических изменений у здоровых испытуемых, а Дельяни, Камурати и Милане, вводившие возрастающие дозы серотонина 10 больным шизофренией как эндолюмбально, так и субокципитально, наблюдали преимущественно вегетативные изменения, иногда увеличение содержания сахара в крови и спинномозговой жидкости; все же у части больных отмечено и преходящее улучшение психического состояния. Прием больших доз 5-ГТФ не дал достаточно выраженного центрального эффекта, как у психически здоровых, так и у больных, хотя и вызывал отчетливые соматические сдвиги. Все же и при приеме 5-ГТФ Тиссо обнаружил у небольшой группы здоровых испытуемых появление на ЭЭГ либо медленных ритмов, либо ?-ритма (у лиц, у которых он отсутствовал на исходной ЭЭГ) развитие сонливости и сна. Таким образом, не удалось обнаружить постоянного и характерного влияния серотонина на психику человека, однако в. части случаев серотонин и 5-ГТФ изменяют психическое состояние, причем складывается впечатление, что психически больные более чувствительны к центральному действию серотонина, чем психически здоровые. Там где удается повлиять на психическое состояние больных введением серотонина, эффект его неоднороден. Как и в экспериментах на животных, отмечены как седативное, так и стимулирующее действие. Чем объясняется разница в чувствительности к серотонину различных людей и разный характер действия серотонина, остается невыясненным. Из числа антагонистов или антиметаболитов[6] серотонина, количество которых чрезвычайно велико, некоторые были испытаны в клинике и обнаружили центральное действие. Так бензил-аналог серотонина, BAS, испытывавшийся первоначально при гипертонической болезни, оказывает наряду с гипотензивным и успокаивающее действие. Его применение как средства лечения психических заболеваний не получило широкого распространения, но все же отдельные исследователи с успехом применяли BAS при шизофрении. Джилле из 26 больных шизофренией с неблагоприятным прогнозом добился улучшения у 8 больных, у 2 из них наступило практическое выздоровление. Однако состояние 5 больных ухудшилось. Улучшение у части больных шизофренией, получавших BAS, отметил также Бенасси. Вассилиу, назначавший другой антагонист серотонина — патокол — 6 беспокойным больным шизофренией, нашел, что все больные стали спокойнее, их поведение — более упорядоченным, а психопатологическая симптоматика менее выраженной. В то же время синтетический антиметаболит серотонина — медмейн — вызывал у людей состояние возбуждения и галлюцинации. Это противоречие Вулли объясняет тем, что антиметаболиты серотонина (как, впрочем, и антиметаболиты любого другого вещества) могут действовать двояко — оказывать серотониноподобное действие, как бы увеличивая содержание серотонина в ЦНС, или вести к недостатку серотонина, блокируя его рецепторы. Эффект в каждом из этих случаев будет противоположным. Однако, пока отсутствуют доказательства того, что влияние на поведение антагонистов или антиметаболитов серотонина обусловлено их влиянием на обмен серотонина, а не какими-либо другими механизмами. Роль серотонина исследовалась также путем изучения изменений его обмена под влиянием ряда лекарств, оказывающих влияние на психику человека. Мы рассмотрим более подробно данные о взаимодействии серотонина с теми лекарствами, психические нарушения при применении которых описаны в настоящей книге. Впрочем, это как раз те вещества, влияние которых на обмен серотонина исследовалось наиболее широко. Обмен серотонина и резерпин. В экспериментах на животных рядом исследователей было показано, что срок появления резерпина в головном мозгу и сроки его выведения из организма, в том числе из ЦНС, не совпадают с временем появления и продолжительностью седативного действия резерпина. В то же время оказалось, что резерпин ведет к уменьшению содержания серотонина в головном мозгу (по данным некоторых авторов — более значительному, чем в других органах) и что кривая падения концентрации серотонина и последующего его накопления совладает по времени с клиническим эффектом резерпина. Содержание свободного серотонина в головном мозгу достигает минимума через несколько часов, через 48 час. оно начинает постепенно повышаться, достигая исходных величин через 7 суток после однократного введения резерпина (опыты Броуди и сотр. на кроликах). Однако исчезновение седативного действия резерпина происходит раньше, чем восстанавливается исходный уровень серотонина, — уже к тому времени, когда содержание связанного серотонина, который под влиянием резерпина почти полностью выводится из ЦНС, начинает повышаться и достигает 15% по отношению к исходной величине. Если резерпин вводят регулярно в течение ряда дней, восстановление содержания серотонина не происходит в течение всего этого периода и начинается только после прекращения приема резерпина. Все время, однако, продолжается выведение с мочой основного метаболита серотонина — 5-ОИУК. Это доказывает, что резерпин не препятствует синтезу серотонина (в этом случае должно было бы произойти практическое исчезновение серотонина из ЦНС и, следовательно, прекращение уже через несколько дней выделения 5-ОИУК), а лишь высвобождает запасы связанного и препятствует связыванию вновь образующегося серотонина. Одна молекула резерпина ведет к высвобождению большого числа молекул серотонина, что, по мнению Броуди и его сотрудников, свидетельствует об изменении под влиянием резерпина состояния участков ЦНС, в которых происходит связывание серотонина. Каков характер этих изменений остается неясным. Ряд других наблюдений подтверждает связь между седативным действием резерпина и нарушением связывания серотонина. Резерпин оказывает слабое седативное действие на мышей и высвобождение серотонина у них незначительно, тогда как у кроликов резерпин вызывает более значительное высвобождение серотонина и оказывает более отчетливое седативное действие (опыты Плечера и Шора). При низкой (0°) и высокой (+37°) температуре окружающей среды высвобождение серотонина не происходит и седативное действие резерпина отсутствует (наблюдения Гараттини и Вальцелли на крысах). Ряд производных раувольфии, снижающих содержание серотонина в головном мозгу, оказывает и седативное действие (десерпидин, ресцинамин), тогда как производные той же раувольфии, как напр., изорезерпин, не оказывающие влияния на обмен серотонина, лишены и седативного эффекта. В то же время препараты, отличающиеся от резерпина по строению, но вызывающие высвобождение серотонина из ЦНС, обладают седативным действием. К таким препаратам относятся производные бензохинолизина, один из которых — нитоман или тетрабеназин — последние годы довольно широко применяется за рубежом для лечения различных психических заболеваний. В опытах на животных нитоман вызывает падение содержания серотонина (и норадреналина) в головном мозгу, не изменяя их содержания в других органах и тканях. Как продолжительность падения концентрации серотонина, так и продолжительность седативного действия нитомана значительно меньше по сравнению с резерпином. К более косвенным доказательствам относится сходство изменений поведения (успокоение, уменьшение моторной активности, потенцирование действия снотворных) и ЭЭГ, вызываемых серотонином и резерпином. Таким образом, гипотеза о связи седативного действия резерпина с изменениями обмена серотонина может быть изложена следующим образом. Резерпин, не влияя на синтез серотонина в головном мозгу, препятствует связыванию образовавшегося серотонина, а также высвобождает значительную часть уже имевшегося запаса связанного серотонина, переводя его в свободное состояние. Свободный серотонин расщепляется моноаминоксидазой и в виде 5-ОИУК выводится из организма. В результате в головном мозгу уменьшается как общая концентрация серотонина, так и содержание свободного серотонина и отношение связанного серотонина к свободному. Седативное действие обусловлено падением концентрации, главным образом, свободного серотонина (по последним данным Броуди и сотр. — нарушением баланса между синтезом и распадом серотонина и изменением содержания серотонина у его рецепторов). Резерпин уменьшает также концентрацию серотонина в других органах и тканях, но как показали опыты с нитоманом, для получения седативного эффекта достаточно избирательного нарушения обмена моноаминов на уровне ЦНС. Резерпин наряду со снижением содержания серотонина вызывает и снижение содержания катехоламинов — адреналина и норадреналина. При этом кривая падения и восстановления содержания катехоламинов в головном мозгу оказалась в опытах на животных почти идентичной с соответствующей кривой серотонина. Отсюда возникло предположение, что основную роль в развитии седативного эффекта после введения резерпина играют изменения обмена катехоламинов. В пользу этого предположения свидетельствуют опыты, в которых допамин (предшественник адреналина) временно устранял седативное действие резерпина, тогда как 5-ГТФ не оказывал никакого эффекта, хотя сочетание допамина с 5-ГТФ вызывало более полное и длительное устранение эффекта резерпина. Гармалин — ингибитор МАО — при введении его после резерпина повышает концентрацию серотонина в головном мозгу, когда концентрация катехоламинов еще остается низкой; седативное действие резерпина при этом не исчезает. Вместе с тем значительное количество наблюдений говорит против предположения о решающей роли снижения концентрации катехоламинов в механизме седативного действия резерпина. Морфин уменьшает у кошек содержание катехоламинов в ЦНС, не влияя на обмен серотонина, — при этом у них развивается состояние возбуждения; холод, препятствующий седативному действию резерпина, не влияет на вызываемое резерпином падение содержания катехоламинов. В последние годы был получен препарат ?-метилметатирозин, избирательно снижающий содержание катехоламинов в головном мозгу. При даче этого препарата животным седативное действие не наступало, а при добавлении резерпина, снижавшего содержание серотонина развивался седативный эффект (опыты Броуди и сотр. на кроликах). Таким образом, все полученные в настоящее время в экспериментах на животных данные не могут быть объяснены ни с точки зрения об исключительной роли изменений обмена серотонина, ни с точки зрения гипотезы об исключительной роли обмена катехоламинов в механизме седативного действия резерпина. Все же большинство экспериментов говорит о том, что более важную роль играют нарушения обмена серотонина. Нельзя, разумеется, исключить предположение, что и другие, еще неизвестные механизмы, помимо нарушения обмена моноаминов, участвуют в седативном действии резерпина. Снижение судорожного порога, неоднократно отмечавшееся как в опытах на животных, так и в клинике (большая тяжесть судорожных припадков, иногда — смертельный исход при применении ЭСТ больным, получающим резерпин, судорожные припадки во время лечения резерпином), также предположительно связывают с влиянием резерпина на обмен серотонина. Однако, как отмечалось выше, данные о противосудорожном действии серотонина (и, следовательно, облегчении возникновения судорожных припадков при снижении концентрации серотонина) недостаточно однородны и противосудорожное действие серотонина не может считаться доказанным. Отметим, что некоторые возражения против гипотезы о связи центрального действия резерпина с обменом серотонина основываются на наблюдениях, показывающих отсутствие полного совпадения между влиянием на ЦНС, оказываемым серотонином и резерпином, — например анальгетическое действие серотонина и отсутствие анальгетического действия резерпина, ослабление под влиянием резерпина потенцирующего действия серотонина на сон, вызванный барбитуратами и т. п. Эти возражения, как нам кажется, основаны на недоразумении — ведь гипотеза эта предполагает, что в основе действия резерпина лежит нарушение обмена серотонина, а вовсе не то, что серотонин и резерпин оказывают одинаковое воздействие на ЦНС. Влияние резерпина на обмен серотонина у человека изучали почти исключительно с помощью определения выделения с мочой 5-ОИУК. Выделение 5-ОИУК возрастает только в первые дни после начала лечения резерпином, иногда значительно: на 25—50%. У части исследованных — преимущественно психически больных — эти изменения отсутствовали. Вслед за начальным подъемом, выделение 5-ОИУК нормализуется, не обнаруживая тенденции ни к дальнейшему нарастанию, ни к чрезмерному падению. Обычно количество 5-ОИУК в суточной моче существенно не отличается от цифр, полученных у контрольных групп — не получающих резерпина — или у тех же больных до начала лечения. Эти факты не проливают никакого дополнительного света на вопрос о влиянии резерпина на обмен серотонина, но и не противоречат гипотезе о существовании такой связи. В самом деле, увеличения выделения 5-ОИУК можно ожидать только в первые дни лечения, когда происходит высвобождение и расщепление запасов связанного серотонина и то лишь в том случае, если этот процесс происходит быстро и ведет к высвобождению больших количеств серотонина. В дальнейшем, поскольку ни синтез, ни разрушение серотонина не нарушены, выделение 5-ОИУК, очевидно, должно оставаться в пределах нормальных цифр, что и отмечается в большинстве исследований такого рода. С другой стороны только начальное усиление выведения 5-ОИУК подтверждает правильность изложенной гипотезы, тогда как сохранение нормальных цифр содержания 5-ОИУК в моче само по себе, разумеется, не указывает на нарушение связывания серотонина в головном мозгу. Во всех перечисленных нами выше фактах и гипотезах речь шла об объяснении обычного — терапевтического действия резерпина. Еще менее определены данные о причинах осложнений, в том числе психических нарушений, возникающих во время лечения резерпином. Судорожные припадки, вызываемые резерпином, предположительно связывают с падением содержания серотонина, что отчасти подтверждают опыты на животных, в которых обнаружено антагонистическое действие серотонина и резерпина по отношению к судорогам, вызываемым метразолом, а также увеличение содержания в головном мозгу серотонина после приема противосудорожного средства — дифенина (дилантина). Однако, как указывалось выше, данные о противосудорожном действии серотонина неоднородны. Неясным остается с этой точки зрения отмеченный в клинике факт появления судорожных припадков только в первые недели лечения и их прекращение, несмотря на продолжение лечения (хотя содержание серотонина в головном мозгу при продолжении лечения должно — судя по опытам на животных — оставаться низким). Состояния возбуждения, нередко возникающие в первые дни лечения резерпином (так называемая турбулентная фаза), могут быть объяснены избытком серотонина, связанным с высвобождением связанного серотонина, который не успевает расщепиться моноаминоксидазой. Депрессивные состояния также могут быть связаны с недостатком серотонина — это согласуется с антидепрессивным действием ингибиторов МАО, увеличивающих содержание серотонина в головном мозгу (см. ниже). Однако только снижением концентрации серотонина в головном мозгу нельзя объяснить развитие депрессивных состояний во время лечения резерпином. Снижение содержания серотонина, если оно лежит в основе седативного действия резерпина, должно происходить у всех или у большинства больных, получающих резерпин. Депрессии же являются осложнением, наблюдающимся у небольшой части больных. Механизм других психических нарушений, возникающих во время лечения резерпином, не удается объяснить нарушениями обмена серотонина. Все эти предположения нуждаются в дальнейшей проверке, так как в настоящее время доказательства их основываются почти исключительно на перенесении данных, полученных в эксперименте на животных, в клинику психических заболеваний. Обычно в литературе можно встретить указание, что хлорпромазин (аминазин) и другие производные фенотиазина, действие которых на психику сходно с действием резерпина, не влияют на обмен серотонина. Это не опровергает гипотезы о связи седативного действия резерпина с нарушением обмена серотонина, т. к. седативное действие может быть обусловлено различными механизмами (по данным Броуди все лекарства, значительно снижающие содержание серотонина в ЦНС, обладают и седативным действием, но это не значит, что снижение концентрации серотонина является единственным возможным механизмом всякого седативного действия). Однако это указание, часто упоминаемое как не вызывающий сомнения факт, находится в противоречии с рядом экспериментальных данных, указывающих на то, что хлорпромазин оказывает влияние на обмен серотонина. В опытах на изолированных органах (отрезок кишки или матка крысы) аминазин является антагонистом серотонина не менее сильным, чем резерпин. Отмечено также антагонистическое действие аминазина и серотонина на сон, вызванный барбитуратами, на артериальное давление у спинальных животных, на условные слюнные и двигательные рефлексы, уменьшение под влиянием аминазина накопления серотонина в головном мозгу, вызываемого ипрониазидом (ипразидом); параллелизм, хотя и не полный, между успокаивающим действием ряда производных фенотиазина и выраженностью их антагонизма к серотонину. Интересны опыты на крысах, проведенные Фридменом и Джайерменом. Они показали, что хлорпромазин, не изменяя общее содержание серотонина в головном мозгу, уменьшает количество связанного и увеличивает количество свободного серотонина. В результате отношение связанного серотонина к свободному снижается до 1,2 по сравнению с 2,5 у контрольных животных. Резерпин, значительно снижающий содержание в мозгу как связанного, так и свободного серотонина, вызывал такое же снижение их соотношения — до 1,4. Фенобарбитал (люминал) в этих экспериментах в противоположность резерпину значительно повышал содержание как связанного, так и свободного серотонина, однако их отношение снижалось в такой же степени, как и у животных, получавших резерпин, и составляло 1,4. Возможно, что седативное действие, которое оказывают и хлорпромазин, и резерпин, и люминал, обусловлено не изменением общего содержания серотонина в головном мозгу, а снижением коэффициента, выражающего отношение связанного серотонина к свободному. О влиянии хлорпромазина на обмен серотонина свидетельствует и наблюдение Коула, отметившего у больной карциноидом тонкого кишечника значительное уменьшение выделения с мочой 5-ОИУК после назначения хлорпромазина. После отмены лечения выделение 5-ОИУК вновь возросло. Имеются сведения, что, по крайней мере, некоторые из производных бутирофенона, получившие распространение в последние годы и оказывающие седативное или, пользуясь более распространенным сейчас термином, — нейролептическое действие, являются антагонистами серотонина. Все эти данные недостаточны для того, чтобы утверждать, что влияние на обмен серотонина является основным или даже существенным звеном в механизме терапевтического действия хлорпромазина, однако они не позволяют безоговорочно отбрасывать такую возможность, как это нередко делается. Серотонин и ингибиторы моноаминоксидазы (МАО). Уже в самом объединении группы препаратов с антидепрессивным действием, клиническое применение которых описано в главе 11-ой 1-й части этой книги, под общим названием ингибиторов МАО показывает, что основное значение в механизме действия этих лекарств придается их способности тормозить МАО. Различные по своему химическому строению препараты, объединенные лишь одним общим свойством — способностью тормозить МАО, оказывали сходное действие на поведение животных и человека, тогда как вещества, близкие к некоторым из ингибиторов МАО, но не блокирующие МАО, не обнаруживали такого эффекта. Если гипотеза о связи антидепрессивного эффекта с торможением МАО пока остается гипотезой, то она уже сейчас дала практический эффект. Пожалуй, впервые в истории психиатрии производились поиски лекарств, которые должны были обладать определенным свойством — способностью блокировать МАО, а заранее предсказанный терапевтический эффект — антидепрессивное действие — подтвердилось при их клиническом испытании. Ряд исследований показал, что имеется параллелизм между клинической эффективностью ингибиторов МАО как антидепрессивных средств и их способностью тормозить действие МАО в экспериментах на животных. Следует иметь в виду, что влияние на МАО может изменяться в зависимости от способа введения препарата. Так например, фенелзин при подкожном введении действует преимущественно на МАО головного мозга, при даче внутрь — преимущественно на МАО печени и т. д. Действие на ряд других ферментов — диаминоксидазу, неспецифические оксидазы, декарбоксилазы, дифосфопиридиннуклеотидазы не обнаруживает корреляции с клиническим эффектом. Поскольку МАО является одним из главных ферментов, обеспечивающих разрушение (путем окислительного дезаминирования) основных моноаминов, оказывающих влияние на ЦНС — серотонина и катехоламинов, естественно возникло предположение, что эффект ингибиторов МАО в клинике связан с накоплением в головном мозгу моноаминов, разрушение которых она блокирует. В ряде экспериментов как in vitro, так и in vivo удалось показать, что под влиянием различных ингибиторов МАО действительно происходит значительное увеличение содержания моноаминов в головном мозгу. Влияние ингибиторов МАО на поведение как экспериментальных животных, так и человека противоположно действию нейролептических средств, в том числе и резерпина — наблюдается оживление моторики, усиление активности, в вегетативной симптоматике — сдвиг в сторону симпатотонии. Седативное действие резерпина извращается, если его сочетают с ингибиторами МАО; этот эффект более отчетлив, если ипрониазид, наиболее часто испытывавшийся в эксперименте (аналогичные данные получены и при применении других ингибиторов МАО), вводят предварительно — до введения резерпина. В этих случаях вместо обычного седативного действия резерпина у животных развивается двигательное возбуждение, пилоэрекция, мидриаз, экзофтальм. Иногда извращение эффекта резерпина не наступает, но и в этих случаях ипрониазид замедляет появление седативного действия резерпина и уменьшает его выраженность. Ингибиторы МАО обнаруживают также антагонизм по отношению к действию резерпина на судорожные припадки, вызываемые электрошоком и метразолом. При комбинации резерпина с ипрониазидом артериальное давление быстро возвращается к исходным цифрам, тогда как резерпин нередко ведет к необратимому падению артериального давления у животных после судорожного припадка. Повышение судорожного порога, вызываемое 5-ГТФ, более значительно, если 5-ГТФ вводят в сочетании с ипрониазидом. В случаях, где у животных, получивших ипрониазид и резерпин, развивается состояние возбуждения, удается отметить совпадение во времени между развитием возбуждения и максимальной концентрацией моноаминов в головном мозгу. Поэтому извращение эффекта резерпина объясняют следующим образом: резерпин вызывает высвобождение моноаминов, находящихся в связанном состоянии, и препятствует связыванию вновь образующихся моноаминов, а ингибиторы МАО препятствуют разрушению моноаминов. В результате резко увеличивается содержание, прежде всего, свободных моноаминов, что и ведет к состоянию возбуждения. Такие же состояния удавалось вызвать у животных с помощью введения больших доз серотонина или его предшественника — 5-ГТФ. Возбуждение развивается и у животных, не получавших резерпина, когда им вводят серотонин или 5-ГТФ (а также норадреналин или его предшественники) на фоне ингибиторов моноаминоксидазы. В этих случаях у части животных развивается не состояние гиперактивности, а успокоение или даже сон, но и эти наблюдения не противоречат предположению о роли изменений содержания моноаминов, так как серотонин, как упоминалось выше, в небольших концентрациях оказывает седативное действие. Таким же образом можно понять и способность ипрониазида и других ингибиторов МАО потенцировать сон, вызываемый барбитуратами или хлоралгидратом. Возможно, однако, что потенцирование действия снотворных средств связано и с тем, что ипрониазид блокирует разрушающие их ферменты и тем самым пролонгирует действие снотворных. Как в эксперименте, так и в клинике можно нередко отметить 2 фазы действия ипрониазида — седативное действие, которое затем сменяется повышением активности и возбуждением. При этом возбуждение по наблюдениям Шора, Броуди и сотр. возникает тогда, когда содержание в головном мозгу моноаминов увеличивается в 2 раза по сравнению с их исходным уровнем. Эта двухфазность действия ингибиторов МАО также может быть объяснена с точки зрения гипотезы о их влиянии на обмен моноаминов — незначительное повышение содержания моноаминов (в первую очередь, серотонина) вызывает седативное действие и лишь по мере дальнейшего их накопления появляется стимулирующий эффект. Более трудным является вопрос о том, накопление какого из моноаминов — серотонина или норадреналина является основным фактором, ведущим к развитию возбуждения. Этот вопрос, как и вопрос о том, является ли падение содержания серотонина или норадреналина причиной седативного действия резерпина, не может считаться окончательно разрешенным. Однако, если большинство экспериментов свидетельствует в пользу связи между седативным эффектом и снижением содержания серотонина, то в отношении возбуждения — наоборот, есть больше оснований связывать его с накоплением катехоламинов. Адренолитическое средство фентоламин купирует возбуждение, вызванное сочетанием ингибиторов МАО и резерпина, такой же эффект оказывает хлорпромазин, в основе успокаивающего действия которого, по мнению большинства исследователей, также лежит его антагонизм к катехоламинам. Напротив, антагонист серотонина, препарат UML-491 в этих случаях неэффективен. Весьма показательную серию экспериментов на мышах и кроликах провела группа Броуди. Они вызывали у животных состояние возбуждения, вводя им на фоне действия ингибитора МАО ?-метилметатирозин, высвобождающий связанный норадреналин, но почти не влияющий на обмен серотонина. В этих условиях происходило, следовательно, накопление только свободного норадреналина. При введении резерпина после ингибиторов МАО возбуждение было выражено значительно слабее, чем после комбинации ингибиторов МАО с ?-метилметатирозином. Дополнительное высвобождение серотонина, таким образом, уменьшало возбуждение, обусловленное норадреналином. Если путем предварительного введения ?-метилметатирозина снижали содержание в ЦНС норадреналина, то последующее введение комбинации ингибиторов МАО и резерпина, при которой возрастало только содержание свободного серотонина, не вызывало возбуждения и даже оказывало преходящее седативное действие. В ряде специальных опытов было показано, что сам ?-метилметатирозин не оказывает стимулирующего влияния на ЦНС. В некоторых экспериментах, однако, удавалось вызвать у животных состояние повышенной двигательной активности, вводя им серотонин или 5-ГТФ. При описании действия серотонина на животных мы упоминали, что большие дозы серотонина или 5-ГТФ сами — и без применения ингибиторов МАО могут вести к состояниям возбуждения. Поэтому нельзя полностью отвергнуть участие серотонина в механизме развития МАО. В последние годы Леви высказала предположение, что как серотонин, так и норадреналин оказывают седативное действие, которое усиливается под влиянием ингибиторов МАО. Возбуждение же связано с накоплением в ЦНС предшественников моноаминов — 5-ГТФ и диоксифенилаланина вследствие блокирования действия карбоксилаз. Наконец, было показано, что ингибиторы МАО увеличивают содержание в организме человека другого производного триптофана — триптамина, который оказывает стимулирующее действие на ЦНС. Поэтому в настоящее время нельзя с определенностью сказать, какие именно изменения обмена в головном мозгу, вызываемые ингибиторами МАО, являются причиной их стимулирующего и антидепрессивного действия. Наиболее полно изучено влияние ингибиторов МАО на обмен моноаминов, но этого еще недостаточно для того, чтобы утверждать, что эти изменения являются и наиболее важными. Клиническое описание изменений, вызываемых ингибиторами МАО у психически здоровых и психически больных, приведено в главе 11-ой. Лабораторные исследования, проведенные на людях и по необходимости ограничивавшиеся исследованиями мочи и — в меньшей степени — крови, не дали существенных данных, проливающих свет на механизм действия ингибиторов МАО. Клинически, как и в эксперименте, отмечено стимулирующее действие, иногда — извращение седативного эффекта резерпина и усиление терапевтического эффекта резерпина с помощью ингибиторов МАО. Бюссов и его сотр. в ряде работ показали, что извращение седативного действия резерпина и других нейролептических средств может быть достигнуто назначением доридена (глютетимида; снотворное средство, обладающее также антихолинергическим действием) и ряда других препаратов, прежде всего антихолинергических — атропина, акинетона, орфенадрина и др. — и симпатомиметических средств, большинство которых не тормозит МАО и, по-видимому, не оказывает влияния на обмен серотонина. Не отмечено и постоянной зависимости между клиническим эффектом ингибиторов МАО и изменениями выделения с мочой 5-ОИУК, основного метаболита серотонина. Вместе с тем, исследования, проведенные на здоровых людях, показали, что характер реакции на введение адреналина и норадреналина — изменения пульса, артериального давления, содержания в плазме адреналина и норадреналина — после предварительного приема ипрониазида остается таким же, как и у людей, которым адреналин и норадреналин вводили без предварительной дачи ипрониазида. Резник и сотр., Киршнер и сотр. показали, что как у здоровых людей, так и у психически больных выделение продуктов обмена адреналина (в частности, метаболизм меченого адреналина) не изменяется под влиянием ипрониазида: блокирование окислительного дезаминирования ведет к компенсаторному усилению другого пути обмена катехоламинов — О-метилирования. В результате, в моче лиц, получивших ипрониазид, увеличивается удельный вес метилированных продуктов обмена катехоламинов, но инактивирование их происходит в обычных пределах и накопления катехоламинов в организме не происходит. Эти данные противоречат гипотезе о связи клинического эффекта ингибиторов МАО с накоплением свободных катехоламинов. Правда, в этих исследованиях речь идет об общем изменении обмена адреналина и норадреналина, тогда как эффект ингибиторов МАО может быть связан с избирательным торможением этого фермента в ЦНС (такое избирательное действие ряда ингибиторов МАО показано в эксперименте на животных). Между тем, метилтрансфераза — опять-таки по данным эксперимента — играет важную роль в расщеплении катехоламинов на периферии и мало влияет на их обмен в ЦНС. По данным Коста и сотр., блокирование метилтрансферазы не ведет к накоплению катехоламинов в головном мозгу, как это наблюдается при блокировании МАО. Несмотря на то, что антидепрессивное действие ингибиторов МАО по мнению большинства исследователей связано с их способностью блокировать действие МАО, не все симптомы, наблюдающиеся в клинике, являются непосредственным следствием этого эффекта. В частности, не доказана связь артериальной гипотензии и токсического действия ряда этих препаратов на печень с торможением МАО. Возможно, влияние ингибиторов МАО на другие органы и системы, которому до сих пор уделялось мало внимания (за исключением терапевтического эффекта при грудной жабе), имеет значение и в клинике вызываемых ими психических изменений. В частности, де Майо нашел, что фенелзин увеличивает активность щитовидной железы как у человека, так и у животных, а влияние желез внутренней секреции, в том числе, щитовидной железы, на психику человека достаточно хорошо известно. Суммируя все приведенные данные, можно сказать, что, хотя механизм действия ингибиторов МАО не может считаться полностью выясненным, имеется достаточно оснований связывать его с торможением МАО, а эффект этого последнего — с повышением тонуса симпатических центров головного мозга, наиболее вероятно — с повышением содержания в головном мозгу норадреналина. Роль изменений обмена серотонина менее вероятна. Частные механизмы, обусловливающие развитие психических нарушений при применении ингибиторов МАО, сколько нам известно, не исследованы, существует лишь общее предположение, что и они связаны, прежде всего, с нарушением обмена моноаминов в головном мозгу. Серотонин и ДЛК (диэтиламид лизергиновой кислоты). Предположение о том, что нарушения обмена серотонина играют важную роль в механизме психотомиметического действия ДЛК, основаны почти исключительно на данных, полученных в эксперименте на животных. В опытах на изолированных органах — участке тонкого кишечника, матке крысы, ухе кролика — был обнаружен антагонизм ДЛК по отношению к серотонину. Однако дальнейшие эксперименты показали, что антагонизм, обнаруженный в опытах на изолированных органах, еще не доказывает наличия такого же антагонизма в целом организме. Вещества, устраняющие действие серотонина на ЦНС, не являются его антагонистами в опытах на периферических органах, а антагонисты серотонина в опытах на изолированных органах оказывают совершенно различное действие на поведение. Так, антагонистами серотонина, наряду с ДЛК, являются бром-ДЛК, обладающий слабым седативным действием, и хлорпромазин — мощное нейролептическое средство. Антагонизм серотонина и ДЛК был обнаружен и в опытах на животных in vivo. Серотонин предотвращает действие ДЛК на хроматофоры рыб, ДЛК блокирует нарушения дыхания и судороги, вызываемые у морских свинок серотонином, отмечено антагонистическое влияние серотонина и ДЛК на прессорные реакции у крыс, на сосуды почек, мускулатуру бронхов, полисинаптические рефлексы кошек. Наибольший интерес представляют наблюдения, свидетельствующие об антагонистическом действии серотонина и ДЛК на уровне ЦНС. К таким экспериментам относятся смена картины депрессии на ЭЭГ после введения серотонина ее активированием при введении ДЛК, устранение под влиянием ДЛК тормозящего действия серотонина на условный рефлекс бегства у крыс и мышей и вызываемого серотонином потенцирования гексобарбиталового наркоза у крыс и мышей, устранение каталептического действия серотонина на собак и кошек. ДЛК тормозил способность митохондриев головного мозга крысы связывать серотонин. При обратном порядке введения серотонин подавлял вызванную ДЛК реакцию пробуждения у мышей, уменьшал действие ДЛК на моторные навыки и спонтанную двигательную активность собак. Вулли сообщает, что в опытах на крысах только интрацеребральное введение серотонина предотвращало влияние ДЛК на поведение животных, тогда как подкожное или внутрибрюшинное его введение эффекта не оказывало, что подтверждает малую проницаемость гемато-энцефалического барьера для серотонина. В морфологических исследованиях показано, что патогистологическое изменения в двигательных ядрах черепно-мозговых нервов и в ретикулярной формации ствола головного мозга, возникающие при продолжительной даче серотонина, выражены значительно слабее, если животным (мышам) дают серотонин в сочетании с ДЛК. Серотонин и ДЛК оказывают одинаковое влияние на нейросекреторную систему гипоталамуса крыс, но при их комбинированном применении эффект не наступает. Однако антагонизм ДЛК и серотонина обнаруживается не во всех случаях или носит частичный характер. Так, ДЛК не изменяет потенцирующего действия серотонина на сон, вызванный мепробаматом (мепротаном), по данным Маррацци и Харта серотонин и ДЛК оказывают одинаковое действие на синапсы, оба вещества усиливают сокращение сердца моллюска. В опытах Шошара и Мазуэ на крысах после введения ДЛК сохранялось действие серотонина на периферическую хронаксию, а после введения серотонина сохраняется действие ДЛК на корковую хронаксию. В гомогенатах головного мозга крыс ДЛК снижает тормозящее действие серотонина на холинэстеразу головного мозга, но не влияет на потенцирующее действие серотонина (серотонин в зависимости от срока инкубации либо тормозит, либо потенцирует действие холинэстеразы). В части опытов ДЛК оказывался то синергистом, то антагонистом серотонина. Это различие иногда зависело от дозы ДЛК, иногда же отмечался 2-фазный эффект — антагонизм ДЛК к серотонину сменялся синергизмом. Такие опыты были проведены как на изолированных органах, так и на животных. Так, например, ритмические сокращения олигодендроглии, наблюдавшиеся Вулли и Шоу (см. выше) состоят из 2-х фаз. В первой фазе наблюдается увеличение и вакуолизация клеток, во второй — их сокращение. По отношению к 1-й фазе серотонин и ДЛК являются антагонистами, по отношению ко второй фазе — синергистами. На основании этих данных было высказано предположение, что ДЛК вызывает нарушения поведения животных и психические нарушения у человека не благодаря антагонизму к серотонину, а вследствие потенцирования его действия В подтверждение этого нередко ссылаются на сходство между нарушениями поведения и вегетативной симптоматикой, вызываемыми у ряда экспериментальных животных ДЛК и большими дозами 5-ГТФ. Эвартс нашел, что у макак ДЛК и близкий по строению к серотонину буфотенин вызывают одинаковые как по картине, так и по длительности изменения поведения. Результаты немногочисленных исследований, проведенных у людей, также дали неоднородные результаты. Усиление и большая продолжительность изменений ЭЭГ, вызываемых серотонином у психически больных, особенно у больных шизофренией, после приема ДЛК (Полони), значительное уменьшение выделения с мочой после приема ДЛК основного метаболита серотонина 5-ОИУК, обнаруженное как у здоровых людей, так и у психически больных Роднайтом и Мак-Илвейном. как будто свидетельствуют о синергизме серотонина и ДЛК. Как серотонин, так и ДЛК в малых дозах потенцируют, а в больших тормозят псевдохолинэстеразу в сыворотке крови человека, что также говорит о сходстве их действия, хотя нет данных, подтверждающих связь между влиянием на холинэстеразу и психотомиметическим эффектом ДЛК. Вместе с тем, списано ослабление эффекта ДЛК при предварительной даче 5-ГТФ (наблюдения Бренджелмена и сотр.), а в некоторых наблюдениях реакция на ДЛК не изменялась ни при комбинации ДЛК с 5-ГТФ (хотя в этих случаях выделение 5-ОИУК с мочой возрастает), ни после приема ДЛК испытуемыми, предварительно получившими резерпин и ипрониазид (т. е. на фоне увеличенного содержания серотонина и катехоламинов в головном мозгу). По данным Масуда у здоровых людей выделение с мочой 5-ОИУК после приема ДЛК не изменяется. Противоречивые результаты дали и попытки изменить реакцию на ДЛК с помощью антагониста серотонина BAS (бензил-аналог серотонина). Клее, Бертино и Каллауэй нашли, что BAS не влияет на эффект ДЛК, а Бенасси отметил ослабление реакции на ДЛК у больных, предварительно получивших BAS. У больных карциноидом тонкого кишечника с высоким содержанием в крови серотонина характер и интенсивность реакции на ДЛК остаются такими же, как у здоровых испытуемых. Это, впрочем, может быть обусловлено тем, что серотонин почти не проникает через гемато-энцефалический барьер. Как на основной аргумент, свидетельствующий против (предположения о связи между психотомиметическим действием ДЛК и его влиянием на обмен серотонина, обычно ссылаются на опыты с бром-ДЛК или препаратом BOL-148. Этот препарат в опытах на изолированных органах, а также в некоторых экспериментах in vivo (по отношению к прессорному эффекту серотонина у крыс, его действию на сосуды почек и мускулатуру бронхов у кошек) оказался более сильным антагонистом серотонина по сравнению с ДЛК. На людях с илеостомией бром-ДЛК тормозил спазм кишки, вызываемый серотонином. Вместе с тем бром-ДЛК не является психотомиметическим средством и даже — напротив — оказывает седативное действие, что доказывает, что он достигает ЦНС и исключает возражение, что препарат оказывает только периферическое действие. Наличие центрального эффекта бром-ДЛК было продемонстрировано и в опытах на животных: бром-ДЛК уменьшал потребление кислорода в гомогенатах головного мозга морских свинок, тормозил синаптическую передачу транскаллозальных импульсов, правда в значительно больших дозах и менее постоянно, чем ДЛК (опыты на кошках), усиливал потенцирующее действие серотонина на вызываемый барбитуратами сон в опытах на крысах. По поводу этого возражения нужно заметить следующее Во-первых, бром-ДЛК является более сильным антагонистом серотонина (чем ДЛК) лишь в опытах на изолированных органах или по отношению к периферическому действию серотонина. Антагонизм к серотонину, обнаруживаемый бром-ДЛК, на уровне ЦНС не продемонстрирован. В приведенных экспериментах бром-ДЛК усиливал действие серотонина (что наблюдалось и в ряде опытов с ДЛК) и его большее, по сравнению с ДЛК, влияние на обмен серотонина в ЦНС не показано. Во-вторых, хотя бром-ДЛК не вызывает столь резких и постоянных изменений психики, как ДЛК, и действует в значительно больших дозах, он может оказывать не только седативное действие, но и вызывать психические нарушения, обнаруживающие известное сходство с реакцией на прием ДЛК. Так, по наблюдениям Бертино, Клин и Вайнтрауба прием 32—64 ?/кг бром-ДЛК вызывал только у 3 из 10 здоровых добровольцев чувство беспокойства и внутренней напряженности, а после приема 128—256 ?/кг у 6 из 10 человек появлялось повышенное настроение, беспечность, иногда — тревога, необычно яркая окраска окружающих предметов, нарушение внимания, которые сохранялись 1—3 часа. По данным Салмоираги и Пейджа, психические нарушения могут возникать и при приеме значительно меньших доз бром-ДЛК — 5—7 ?/кг и усиливаются, если вместе с бром-ДЛК вводят серотонин в дозах, которые сами по себе не влияют на поведение. Значительные дозы бром-ДЛК (1 мг и более) могут усиливать реакцию на ДЛК. Вопрос о развитии перекрестного привыкания не вполне решен, отмечено как появление устойчивости к ДЛК после предварительного приема бром-ДЛК, так и отсутствие такого эффекта. По-видимому, эти разногласия зависят от разницы в дозах как бром-ДЛК, так и даваемых затем доз ДЛК. Наконец, известен случай психоза, весьма сходного с психозом, вызываемым ДЛК, который развился у здорового мужчины после приема 0,5 мг бром-ДЛК. Во время психоза наблюдались смена эйфории и страха, яркость окраски окружающих предметов, зрительные иллюзии, гиперакузия, изменения схемы тела, затрудненное выполнение психологических тестов при сохранной ориентировке в окружающем. Психоз длился около 7 часов с последующей астенией в течение 3 дней (наблюдение Ричардса и сотр.; единственный случай, который мы нашли в литературе). Таким образом, нельзя говорить об отсутствии психотомиметического действия бром-ДЛК. Безусловно, дозы бром-ДЛК, при которых появляются психические нарушения, значительно выше, чем дозы ДЛК (напомним, что психические изменения вызывает уже прием 1—2 ?/кг ДЛК, а иногда и меньше). Однако большинство психотомиметиков оказывает действие в дозах, значительно превышающих дозы ДЛК. Достаточно сказать, что обычная доза мескалина, ведущая к отчетливым изменениям психики, примерно в 5000 раз больше соответствующей дозы ДЛК. По этим основаниям (психотомиметический эффект больших доз бром-ДЛК и отсутствие доказательств более сильного его влияния на обмен серотонина на уровне ЦНС по сравнению с ДЛК) мы полагаем, что опыты с бром-ДЛК не могут опровергнуть роли нарушений обмена серотонина в генезе психических нарушений, вызываемых ДЛК. Разумеется, имеющиеся в настоящее время данные недостаточны для того, чтобы утверждать, что нарушения обмена серотонина являются причиной или одним из основных механизмов психотомиметического действия ДЛК. Большинство доказательств получено в эксперименте на животных, не может быть безоговорочно перенесено на человека и, кроме того, носит косвенный характер. Результаты экспериментов не вполне однородны, хотя больше данных, свидетельствующих в пользу антагонистического действия ДЛК и серотонина на поведение. Этот антагонизм может быть и не специфичным и сводиться к противоположному характеру действия на ЦНС седативных средств, к которым, по-видимому, относится серотонин, и средств возбуждающих, к которым относится ДЛК. Однако роль взаимодействия ДЛК и серотонина остается вероятным предположением, нуждающимся в дальнейшей проверке. Сведения о взаимодействии серотонина с другими психотомиметическими и лекарственными средствами Немногочисленны. Мескалин в опытах на изолированных органах и in vivo (в эксперименте на животных) усиливает эффект серотонина, в частности, потенцирование под влиянием серотонина сна, вызванного барбитуратами. Амизил (бенактизин) является антагонистом серотонина в опытах на изолированных органах, но увеличивает содержание серотонина в гипоталамусе и таламусе экспериментальных животных. На вероятность связи психотомиметического действия бенактизина с нарушениями обмена серотонина указывает наблюдавшееся у испытуемых резкое падение выделения 5-ОИУК на высоте психических нарушений. Данные о влиянии тофранила на обмен серотонина недостаточны. На изолированных органах тофранил является антагонистом серотонина. В опытах на крысах отмечено увеличение содержания серотонина в головном мозгу, при этом по данным Джайермена тофранил увеличивает преимущественно количество связанного серотонина и меньше влияет на содержание свободного серотонина, общее же соотношение связанного и свободного серотонина не меняется. По данным Коста и сотр. тофранил препятствует снижению содержания серотонина, вызываемому резерпином. Эти наблюдения не подтверждаются другими исследователями, которые нашли, что содержание серотонина в головном мозгу снижается при сочетании тофранила с резерпином в такой же степени, как и после введения одного резерпина. Однако по Джайермену отношение связанного серотонина к свободному остается таким же, как и после введения одного тофранила (резерпин, наряду со снижением общего содержания серотонина, снижает и отношение связанного серотонина к свободному). Данные о центральном антагонизме тофранила и серотонина получены только у животных и недостаточно убедительны, в частности, противоречивы данные об антагонизме тофранила и резерпина на уровне ЦНС. У больных, лечившихся тофранилом, отмечено падение содержания серотонина в крови, но этот периферический эффект не может быть использован для объяснения взаимодействия серотонина и тофранила на уровне ЦНС. Данные о влиянии тофранила на МАО также противоречивы. Во всяком случае, имеющиеся в настоящее время данные не позволяют говорить о принципиальном отличии механизма действия тофранила от ингибиторов МАО. В заключение кратко остановимся на вопросе о роли серотонина при психических заболеваниях. Еще в 1954 году Вулли и Шоу высказали предположение, что шизофрения может быть вызвана либо недостатком, либо избытком серотонина. Вполне понятно, что возможность выявления нарушений обмена, лежащих в основе шизофрении, изучение патогенеза которой на протяжении десятилетий не дало ощутимых результатов, привлекла внимание многих психиатров. Большинство исследований обмена серотонина базировалось на определении выделения с мочой 5-ОИУК. Оценка результатов подобных исследований несколько затрудняется тем обстоятельством, что нормальные цифры содержания 5-ОИУК в моче колеблются в довольно широких пределах. Все же почти все исследователи приходят к выводу, что нет существенней разницы в выделении с мочой 5-ОИУК у больных шизофренией по сравнению с больными другими нервными и психическими заболеваниями, а также психически и соматически здоровыми людьми, в том числе после нагрузки серотонином или его предшественником — 5-ГТФ. Не отмечено и существенной разницы в содержании серотонина в крови у здоровых людей и больных шизофренией. Правда, авторы некоторых из этих исследований столкнулись с тем же явлением, с каким сталкивались почти все исследователи, изучавшие ту или иную форму нарушений обмена при шизофрении: диапазон выделения 5-ОИУК и содержания серотонина в крови был у больных шизофренией больше, чем у здоровых; у части больных шизофренией цифры были выше, у части ниже, и наконец, у части больных — такими же, как в контрольных группах. Лечение и его эффект мало влияли на выделение 5-ОИУК с мочой. Результат этих исследований не является неожиданностью. Принцип диагностики шизофрении и отграничения ее от других заболеваний, а, следовательно, и состав больных, у которых производились эти исследования, в слишком большой мере зависят от точек зрения авторов, как известно, весьма существенно отличающихся друг от друга. Если к этому добавить, что формы проявления и стадии болезни могут быть весьма разнообразными, трудно ожидать получения однородных результатов. Все же Дж. Бускаино и Стефанаки, наиболее тщательно исследовавшим выделение 5-ОИУК при различных заболеваниях, с помощью хроматографии на бумаге удалось подметить некоторые особенности обмена серотонина при шизофрении. По их данным оказалось, что средние цифры содержания 5-ОИУК в суточной моче у больных шизофренией несколько выше, чем у здоровых, больных другими психическими, а также неврологическими заболеваниями. Если же выделить группы больных шизофренией в зависимости от формы и давности заболевания, различие становится еще более отчетливым. Так, у здоровых испытуемых суточная моча содержит в среднем 5,15 мг 5-ОИУК, максимально 9,2 мг. У больных шизофренией средняя цифра — 6,1 мг, но максимальная — 26 мг. При этом у недавно заболевших шизофреников средние цифры значительно выше, чем у здоровых — 8,3 мг, а у больных с большой давностью заболевания несколько ниже — 4,2 мг. Наиболее высокие средние цифры — 10,01 мг обнаружены у больных кататоничеокой формой шизофрении. Большая часть исследований выделения 5-ОИУК с мочой у здоровых и психически больных, проводилась однократно и без сопоставления с динамикой психического состояния больного. Значительно больший интерес представляют динамические исследования. Брюн и Химвич обнаружили у небольшой группы больных шизофренией совпадение ухудшения психического состояния с увеличением выделения 5-ОИУК, и успокоения — с падением выделения 5-ОИУК (хотя у 2 больных падение содержания 5-ОИУК не сопровождалось успокоением). Брюн и Пшайдт отметили наряду с усиленным выведением 5-ОИУК увеличение — при ухудшении состояния — содержания в моче триптамина и 3-индолуксусной кислоты. Интересно отметить, что эти изменения метаболизма индолов предшествовали изменению психического состояния больных и не зависели от того, сопровождалось ли ухудшение усилением двигательного возбуждения. Лунгберг пользовался повторными определениями выделения 5-ОИУК с мочой, сопоставляя их динамикой психического состояния больных, и варьировал лечение в зависимости от результатов этих исследований. Он получил при этом значительно лучшие результаты, чем при назначении лечения только на основании особенностей клинической симптоматики. Сходные данные приведены в работе Геллера. Точно так же динамическое исследование содержания серотонина в крови позволило Юсу и сотр. установить известную корреляцию между обменом серотонина и изменениями психического состояния больных. Они отметили, что острое психотическое состояние сопровождалось быстрым падением содержания в крови серотонина, которое по мере улучшения состояния больных постепенно повышалось, но оставалась низким у тех больных, состояние которых не улучшилось. Все эти данные нуждаются в дальнейшей проверке, но они показывают, что нарушения обмена серотонина могут играть роль при психических заболеваниях. Нарушения обмена серотонина отмечены при фенилпировиноградной олигофрении (уменьшение выделения 5-ОИУК), при циркулярной депрессии (повышение содержания серотонина в крови), но все эти данные немногочисленны и с трудом поддаются систематизации, так как усилия большинства исследователей были сосредоточены на изучении шизофрении. Лечебное применение серотонина при шизофрении изучалось в отдельных работах. Обычно оно сочеталось с другими методами лечения, что не позволяет сделать определенных выводов, хотя в литературе можно найти указания на благоприятный эффект лечения шизофрении сочетанием серотонина и нейролептических средств или 5-ГТФ с BAS. Впрочем, вряд ли можно найти лекарство, которое не было бы испробовано при шизофрении и не нашло бы сторонников, высказывавшихся за его применение. При оценке данных об обмене серотонина следует иметь в виду, что серотонин содержится в различных органах и тканях и что количество его в ЦНС составляет весьма незначительную часть всего серотонина, тогда как выделение 5-ОИУК с мочой и содержание серотонина в крови отражают обмен серотонина в организме в целом. Напомним, что только 3—4% 5-ОИУК, обнаруживаемой в моче, образуется за счет метаболизма серотонина в головном мозгу (данные Эрспамера). Понятно поэтому, что даже значительное увеличение или уменьшение обмена серотонина в мозгу не может оказать существенного влияния на общее количество выделяющейся с мочой 5-ОИУК. Поэтому нормальные величины серотонина крови и 5-ОИУК в моче говорят только об отсутствии общих изменений метаболизма серотонина и не исключают возможности избирательного его нарушения в ЦНС. Кроме того, 5-ОИУК является хотя и основным, но не единственным метаболитом серотонина. У здоровых людей около 40% а у больных шизофренией всего около 25% серотонина выводится с мочой в виде 5-ОИУК (Бускаино и Стефанаки). Следовательно, изменения выделения 5-ОИУК не отражают всех изменений обмена серотонина. С другой стороны изменения обмена серотонина могут происходить и при ряде соматических заболеваний, не сопровождающихся психическими нарушениями. Упомянем в качестве примера снижение содержания серотонина в крови у больных, подвергшихся резекции желудка, или при заболеваниях печени. На обмен серотонина и выделение 5-ОИУК может влиять и характер пищи (некоторые фрукты и овощи (бананы, грецкие орехи, помидоры и др.) содержат большое количество серотонина). Одним из существенных возражений против гипотезы о роли серотонина в происхождении психических нарушений являются наблюдения над больными, страдающими карциноидом тонкого кишечника. Несмотря на то, что содержание серотонина в крови у этих больных во много раз выше, чем у здоровых людей, они не обнаруживают никаких психических нарушений, за исключением изменений психики, обычно наблюдаемых при тяжелых соматических заболеваниях. Это может быть связано с тем, что серотонин не проникает через гемато-энцефалический барьер, как это допускают многие исследователи. Повышенное содержание серотонина в крови в этом случае не должно влиять на обмен серотонина в головном мозгу. Большее количество съеденных бананов тоже может резко увеличить содержание серотонина в организме, но психические нарушения при этом, как известно, не наступают. Наблюдения, в которых внутривенное введение серотонина изменяло психическое состояние больных, указывает на то, что в некоторых случаях он может проникать через гемато-энцефалический барьер, но эти наблюдения относятся к психически больным, у которых повышенная проницаемость барьера может быть обусловлена их заболеванием. Возможно также, что увеличение поступления серотонина в ЦНС само по себе не вызывает психических нарушений, если не нарушены ни его связывание тканью мозга, ни расщепление. Лабори, Куаро и сотр. выдвинули гипотезу, согласно которой функциональное состояние нервных клеток зависит не от общего количества свободного или связанного серотонина, а от их соотношения, которое в случае карциноида кишечника не должно меняться. При постепенном и постоянном увеличении содержания серотонина, головной мозг может приспосабливаться к новым условиям, обнаруживая пониженную чувствительность к избытку серотонина. Интересен в этой связи случай, описанный Саузреном, Уорнером и сотр. У наблюдавшейся ими больней, у которой в течение ряда лет отмечались большие судорожные припадки, приступообразные состояния возбуждения и обездвиженности с последующей амнезией, эмоциональная лабильность, атаксия и дизметрия, содержание серотонина в крови было очень высоким (4,0 ?/мл, т. е. в 10—20 раз выше нормы). Дача этой больной в течение 4 дней 1 мг в день резерпина вызвала резкое падение содержания серотонина в крови (до 0,03 ?/мл), увеличение содержания в моче 5-ОИУК, а затем полное ее исчезновение. Одновременно у больной появились покраснение кожи, тремор, возбуждение, чувство, что она сходит с ума, усиление атаксии. Все эти симптомы исчезли после отмены резерпина и восстановления прежнего уровня серотонина в крови. Таким образом, эта больная обнаруживала пониженную чувствительность к избытку серотонина и повышенную — к его потере. Данные экспериментов на животных, как и исследования здоровых и психически больных людей, приведенные в этой главе, указывают на то, что серотонин, являющийся нормальным продуктом жизнедеятельности организма, играет роль и в деятельности ЦНС. На это указывает как избирательная локализация в ЦНС, так и ряд данных, свидетельствующих о центральном действии экзогенного серотонина (при введении его в ЦНС), и об изменении психического состояния человека и животных под влиянием лекарств, изменяющих содержание серотонина в ЦНС. Следует отметить, что даже изменения обмена какого-нибудь одного вещества и действие на это вещество одного лекарственного препарата значительно сложнее, чем это может быть определено в клинике или эксперименте. В качестве иллюстрации этого положения приводим схему Джайермена: 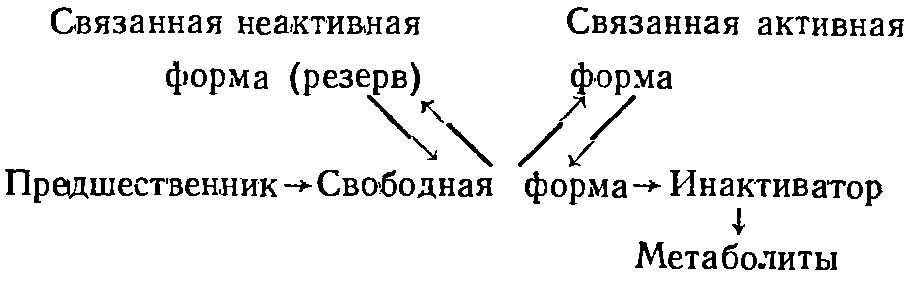 Лекарство может: 1) тормозить или активировать синтез вещества, в данном случае — серотонина, 2) тормозить действие инактиватора, 3) блокировать связь вещества с рецептором, 4) высвобождать вещество из связанного неактивного состояния, 5) усиливать его связывание рецептором или превращение в неактивную форму или то и другое вместе и, наконец, одновременно действовать на несколько из перечисленных механизмов. Понятно поэтому, что оценка обмена по выделению конечных метаболитов, которой обычно приходится ограничиваться при изучении обмена серотонина у человека, не может дать полного представления о действительных изменениях его обмена. Роль, которую играет серотонин в ЦНС, окончательно не установлена. Наиболее распространенной и получившей наибольшее признание, является гипотеза Броуди и его сотрудников, которые считают серотонин нейрогормоном — медиатором трофотропной (парасимпатической) системы в головном мозгу, хотя далеко не все факты удается объяснить с помощью этой гипотезы. Предположение, что нарушение обмена серотонина может сопровождаться психическими нарушениями и что при психических заболеваниях происходит нарушение обмена серотонина, весьма правдоподобно. По-видимому, не случайно многие психотомиметические средства оказались производными триптамина, весьма близкими по строению к серотонину. Возможность образования некоторых из этих веществ — буфотенина и диметилтриптамина (ДМТ) в животном организме недавно показана. Возможно, что причиной психических нарушений является не просто недостаток или избыток серотонина, а образование патологических продуктов его обмена — близких или идентичных с уже известными психотомиметиками — буфотенином, ДМТ, ДЭТ, псилоцином (см. табл.). 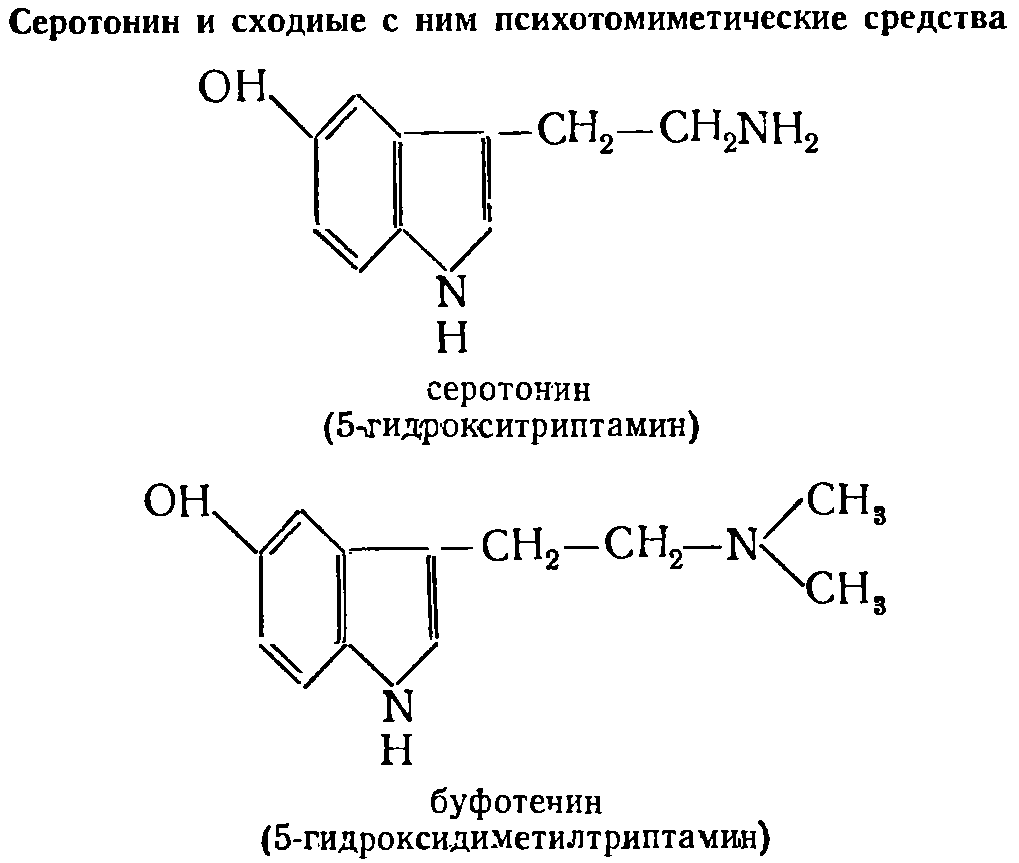 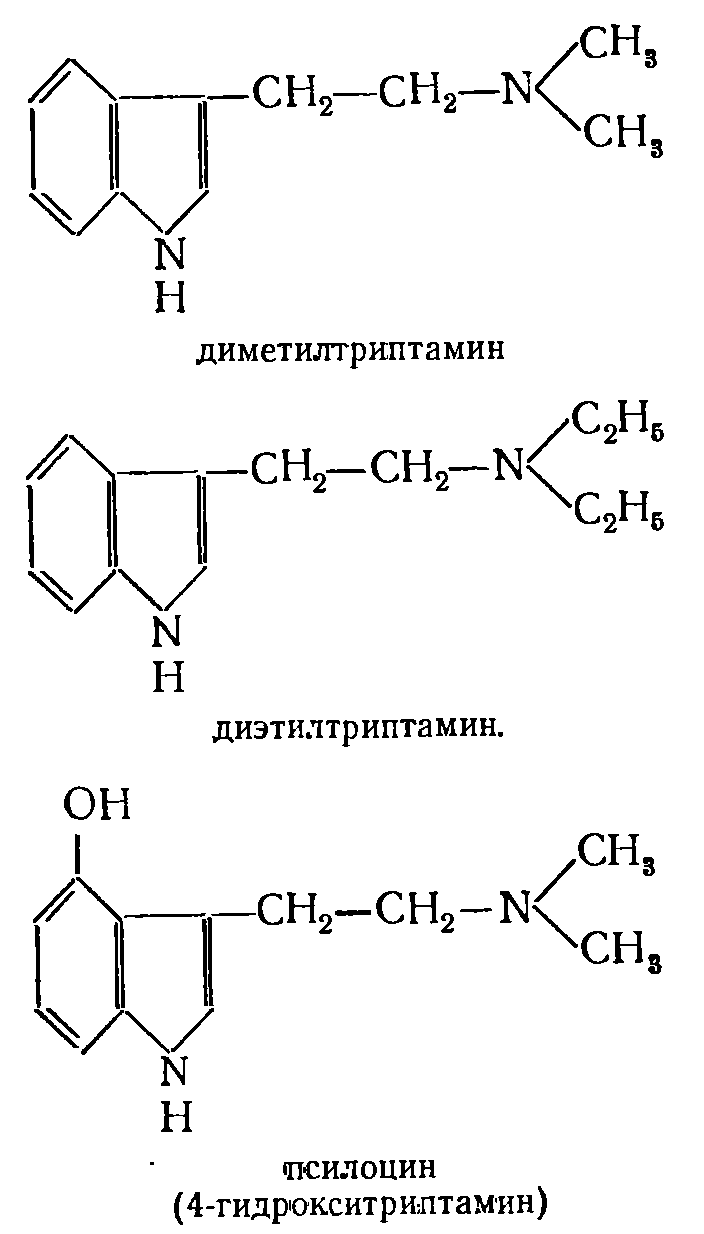 Вместе с тем, мы считаем необходимым подчеркнуть, чтя попытка объяснить патогенез того или иного психического заболевания как следствие нарушения обмена серотонина вряд ли может при нынешнем уровне наших знаний, привести к успешному результату. Тем более, это относится к шизофрении — заболеванию с чрезвычайно пестрой, разнообразной симптоматикой, с разными типами течения и исхода относительно которого даже неизвестно, является ли оно вообще единым заболеванием или группой заболеваний. Большая частота и неблагоприятное в целом течение шизофрении делают психологически вполне понятным стремление психиатров возможно скорее разрешить проблему шизофрении. Это приводит к возникновению большого числа самых разнообразных гипотез, к которым относится и гипотеза Вулли и Шоу о шизофрении как следствии нарушения обмена серотонина. Как и следовало ожидать, эта гипотеза, которая с самого начала была мало обоснованной, не подтвердилась при проверке. В самом деле, Вулли и Шоу исходили из того, что поскольку нарушения обмена серотонина могут вести к психическим нарушениям, а резерпин, применяющийся как средство лечения шизофрении (но вовсе не являющийся специфическим методом ее лечения!) влияет на обмен серотонина, следовательно, нарушение обмена серотонина и является причиной шизофрении. Вывод этот вовсе не вытекает из посылок. Но вред гипотезы Вулли и Шоу заключается не только в том, что она не дала ничего существенно нового для понимания патогенеза шизофрении, но и в этом, что неудачные попытки, предпринятые в этом направлении, вызвали обратную реакцию — стали появляться голоса, полностью отрицающие значение нарушений обмена серотонина при психических заболеваниях, был даже создан термин «сказка о серотонине». Между тем вполне вероятно, что нарушения обмена серотонина — а может быть и других производных триптофана — участвуют в генезе психических заболеваний и что некоторые симптомы или синдромы связаны с этими нарушениями. Именно этот вопрос — вопрос о том, существуют ли определенные состояния или симптомы, при которых нарушен обмен серотонина, каков характер этих нарушений и можно ли их устранить, устраняя дефект обмена серотонина, и должен, по нашему мнению, явиться предметом изучения в настоящее время. Вряд ли будет найдено одно вещество, обмен которого обусловливает шизофрению или какое-либо другое психическое заболевание. Нарушения обмена при психозах, несомненно, более сложны, только при изучении взаимодействия целого ряда факторов может быть достигнуто понимание патогенеза психозов, но среди этих факторов, судя по имеющимся данным, определенное, может быть скромное, место будет принадлежать серотонину. ЛИТЕРАТУРА1. Банщиков В. М. и Столяров Г. В. — Ж. невропат. психиатр., 1961, Т. 61, стр. 934. 2. Ландо Л. И., Захарьин Ю. Л., Крупенина Л. Б. — Ж. невропат. психиатр., 1962, т. 62, стр. 99 3. Машковский М. Д., Рощина Л. Ф. — Ж. невропат. психиатр., 1962, т. 62 стр. 1508. 4. Могилевский А. Я. — Усп. соврем, биол., 1960, вып. 3, стр. 322. 5. Пасхина Т. С. — Вопр. мед. химии, 1960, т. 6, стр. 447. 6. Раушенбах М. О. Чернов Г. А. — Пробл. гематол., 1959, т. 4, стр. 3. 7. Скугаревский А. Ф. — Здравоохр. Белоруссии, 1962, № 6, стр. 45. 8. Старых Н. Т. — X БЭБиМ, 1962, т. 54, стр. 76. 9. Чернов Г. А. — Мед. радиология, 1960, т. 5 стр. 75. 10. Amin A., Crawford Т., Gaddum J. — J. Physiol., 1954, v. 126. p. 596. 11. Angrisani D. — Lav. neuropsichiat., 1961, v. 28, p. 488. 12. Antonell i A., Bertaccini G., Mantegazzini P. — J. Neurochem., 1961, v. 8, p. 157. 13. Arоra R. B. — Life Sci., 1962, № 11, p. 571. 14. Ashcroft G., Sharman D. — Nature, 1960, v. 186, p. 1050. 15. Васiоссhi M., Grandmontagne O., Guillerm H., Ronaux J. — Ann. med. psychol., 1961, v. 1, p. 780. 16. Banerjee S., Agarwal P., — Proc. Soc. exp. Biol. Med., 1958, v. 97, p. 657. 17. Вartlet A. — Brit. J. Pharmacol., 1960, v. 15, p. 140. 18. Вaruk H., Launау J., coll. — Ann. med. psychol., 1958, v. 1, p. 115. 19. Вaruk H., Launау J., coll. — Ann. med. psychol., 1959, v. 117, p. 408. 20. Вein H. — Pharmacol. Rev., 1956, v. 8, p. 435. 21. Вenassi P. — Riv. sperim. freniat., 1958, v. 82, p. 330. 22. Вenassi С, Вenassi P., Allegri G., Bellarin P. — J. Neruochem., 1961, v. 7, p. 264. 23. Benassi P., Bertolotti P. — Riv. sperim. Freniat., 1960, v. 84, p. 351. 24. Вenda Ph., Miravet L. — C. r. Soc. biol., 1957, v. 151, p. 2064. 25. Вenditt E., Rоwleу D. — Science, 1956, v. 123, p. 24. 26. Вerger F., Campbell G., coll. — Ann. N. Y. Acad. Sci., 1957, v. 66, p. 686. 27. Вernaola V. — Rev. Psiquiat. Peru, 1960, v. 3, p. 65. 28. Вertaccini G. — J. neurochem., 1959, v. 4, p. 217. 29. Bertelli A, Gallie G., Genovese E. — Boll. Soc. ital. biol. sperim., 1956, v. 32, p. 822. 30. Вianсhi С — Nature, 1957, v. 179, p. 202. 31. Bleuler М. — Dtsch. med. Wschr., 1956, Bd. 81, S. 1078. 32. Bogdanski D. — J. Pharmac. exp. Therap., 1956, v. 117, p. 82. 33. Bogdanski D., Udenfriend S. — J. Pharmacol. exper. Therap., 1956, v. 116, p. 7. 34. Bogdanski D., Weissbach H., Udenfriend S. — Fed. Proc., 1956, v. 15, p. 402. 35. Bogdanski D., Weissbach H., Udenfriend S., — J. Pharmacol. exper. Therap., 1958, v. 122, p. 182. 36. Bonamini F., Garello L. — Sistema nerv., 1956, v. 8/3, p. 157. 37. Bonnycastle D., Paasonen M., Giarman N. — Nature, 1956, v. 178, p. 990. 38. Вrengelmann J., Pare C., Sandler M. — J. Ment. Sci., 1959, v. 105, p. 770. 39. Brodie B. — Dis. Nerv. Syst., 1960, v, 21. Sect. 2, p. 107. 40. Brodie В., Finger K., Orlans F., coll. — J. Pharmacol. Exper. Therap., 1960, v. 129, p. 250. 41. Brodie В. — Life Sci., 1962, v. 10, p. 551. 42. Brodie В., Olin J., Kuntzman R., Shore P. — Science, 1957, v. 125, p. 1293. 43. Brodie В., Pletsсher A., Shore P. — Science, 1955, v. 122, p. 968. 44. Brodie В., Shоre P. — Ann. N. Y. Acad. Sci., 1957, v. 66, p. 631. 45. Brodie В., Shоre P., Pletsсher A. — Science, 1956, v. 123, p. 992. 46. Brodie В., Sulser F., Costa E. — Rev. Canad. biol., 1961, v. 20, p. 279. 47. Brodie B., Tomich E., Kuntzman R., Shore P. — J. Pharmacol, exper. Therap., 1957, v. 119, p. 461. 48. Bruckman N., Kitchener M., Saunders S., Kline N. — Am. J. Psychiat., 1957, v. 114, p. 262. 49. Brune G., Himwich H. — Science, 1961, v. 133, p. 190. 50. Brune G., Pscheidt G. — Fed. Proc., 1961, v. 20, p. 889. 51. Вulle P. — Proc. Soc exper. Biol. Med., 1957, v. 94, p. 553. 52. Вulle P., Konсhegul L. — J. clin. exper. Psychopathol., 1957, v. 18, p. 287. 53. Buscaino G. A. — Acta Neurol., 1957, v. 12, p. 592. 54. Buscaino G. A. — Acta Neurol., 1961, v. 16, p. 93. 55. Buscaino G. A., Radiсhi M., Stefanachi L. — Acta Neurol., 1958, v. 1, p. 44. 56. Buscaino G. A., Stefanachi L. — Acta Neurol., 1957, v. 12, p. 378. 57. Buscaino G. A., Stefanachi L. — Acta Neurol., 1957, v. 12, p. 156. 58. Buscaino G. A., Stefanachi L. — Acta Neurol., 1957, v. 12, p. 1188 59. Buscaino G. A., Stefanachi L. — Arch. Neurol. Psychiat., 1958, v. 80, p. 78. 60. Buscaino G. A., Stefanachi L. — Confinia neurol., 1958, v. 18, p. 188. 61. Вussow H., Fuhr J. — Nervenarzt, 1958, Bd. 29, S. 268. 62. Сahn J., Herold M., Geоrges G., Pierre R. — Therapie, 1958, v. 13. p. 464. 63. Сampi L., Сamurati C, — Nevrasse, 1956, v. 6, p. 469. 64. Canal N., Maffei-Faccioli A. — J. neurochem., 1959, v. 5, p. 99. 65. Carlsson A., Lindqvist M., Magnusson Т., — Nature, 1957, v. 180, p. 1200 66. Carlsson A., Shore P., Brodie B. — J. Pharm. exper. Therap., 1957, v. 120, p. 334. 67. Chen G., Bohner B. — J. Pharmacol. Exper. Therap., 1961, v. 131, p. 179. 68. Chessin M., Kramer E., Scott Ch. — J. Pharmacol, exper. Therap., 1957, v. 119. p. 453. 69. Cole J., Вertinо G. — Proc. Soc. Exper. Biol. Med., 1956, v. 93, p. 100. 70. Collier G., Martin A. — Ann. med. psychol., 1959, v. 2, p. 491. 71. Consbruch U., Faust CI. — Psychiat. Neurol., 1960, Bd. 140, S. 76. 72. Consbruch U., Faust CI. — Wien. klin. Wschr., 1960, Bd. 72. S. 285. 73. Cook L., Weidley E. — Ann. N. Y. Acad. Sci., 1957, v. 66, p. 740. 74. Coppen A. — Lancet, 1963, v. 1, p. 79. 75. Costa E. — Psychiat. Res. Rep., 1956, v. 4, p. 11. 76. Costa E., Aprisоn M. — J. Nerv. Ment. Dis., 1958, v. 126, p. 289. 77. Costa E., Aprisоn M. — Amer. J. Physiol., 1958, v. 192, p. 95. 78. Costa E., Gessa G., Kuntzman R., Brodie B. — Proc. 1st internat. Pharmacol. Meet. v. 8, Oxford — London — New York — Paris, 1961, p. 47. 79. Costa E., Mоrpurgо C, Revzin A. — Rec. Advanc. Biol. Psychiat., 1961, v. 3, p. 122. 80. Сrepax P., Infantellina F. — Arch. Sci., biol., 1957, v. 41, p. 207. 81. Crome L., Pare С — J. Ment. Sci., 1960, v. 106, p. 862. 82. Cuenca E., Costa E., Kuntzman R., Brodie B. — Med. exptl., 1961, v. 5, p. 20. 83. Darling H., Kruse W., Hess G., Holrmann M. — Dis. Nerv. Syst., 1959, v. 20, p. 269. 84. Degnwitz R., Frowein R., coll. — Klin. Wschr., 1962, Bd. 40, S. 285 85. Delaу J., Piсhоt P., coll. — Presse med., 1962. v. 70, p. 2147. 86. Delmas-Marsalet P. — Rev. Neurol., 1960, v. 102, p. 185. 87. Nоdal J., Tоrtras J. — Med. Clin. (Barcelona), 1957, v. 29, p. 36. 88. Denys A., Levy J., Miche-Ber E. — C. r. Acad sci., 1962, v. 255, p. 2528. 89. Dоgliani P., Сamurati С, Мilano G. — Nevrasse, 1958, v. 6. p. 340. 90. Dolce G., Garello L. — Boll. soc. ital. biol. sperim., 1956, v. 32, p. 441. 91. Donaldson R., Zaidman I., Gray S. — Gastroenterology, 1960 v. 38, p. 937. 92. Ehringer H., Hornykiewicz O., Lechner K. — Arch. exper. Pathol. Pharmakol., 1960, Bd. 239, S. 507. 93. Ernst A., von Andel H., Charbon G. — Psychopharmacologia, 1961, v. 2, p. 425. 94. Erspamer V. — Experienta, 1956, v. 12, p. 63. 95. Erspamer V. — Arzneimittel-Forsch., 1958, Bd. 8, S. 571. 96. Erspamer V., Bertaccini G. — Arch. internat. Pharmacodyn., 1962, v. 137, p. 6. 97. Fabing H. — J. Ment. Sci., 1958. v. 104, p. 573. 98. Fastier F., Speden R., Waal H. — Brit. J. Pharmacol., 1957, v. 12, p. 251. 99. Feldberg W, Sherwood S. — J. Physiol., 1954, v. 123, p. 148. 100. Feldman S., Izak G., Nelken D. — Acta psychiat. neurol. Scand., 1957. v. 32, p. 37. 101. Feldstein A., Freeman H., coll., — Amer. J. Psychiat., 1959, v. 116, p. 219. 102. Feldstein A., Hoagland H., Freeman H. — Science, 1958, v. 128, p. 358. 103. Feldstein A., Hoagland H., Freeman H. — J. Nerv. Ment. Dis., 1959, v. 129, p. 62. 104. Feldstein A., Hoagland H., Freeman H. — Science, 1959, v. 130, p. 500. 105. Feldstein A., Hoagland H., Freeman H. — Arch. gen. Psychiat., 1961, v. 5, p. 246. 106. Ferro Milone F., Gomirato G. — Glorn. psichiatr. neuropatol., 1957, v. 85, p. 519. 107. Fischer R., Griffin F., Liss L. — Ann. N. Y. Acad. Sci.. 1962, v. 96, p. 44. 108. Florn R. — Stud. Cercet. Fiziol., 1958, Bd. 3, S. 311. 109. Florn R., Sterescu-Volanschi M., Costin A. — Rev. sci. med R. P. R., 1960, v. 5, p. 149. 110. Fоrnaroli P., Keller M. — Farmaco ed. sci., 1954, v. 9. p. 546. 111. Fоrrest A. — J. Ment. Sci., 1957, v. 103, p. 614. 112. Fragosa Mendes J., Lopes de Rosario J. — Encephale, 1959, v. 48, p. 501. 113. Freedmann D., Benton A. — N. Eng. J. Med., 1961, v. 264, p. 529. 114. Fresia P., Genоvese E., Katо R., Valzelli L. — Boll. Soc. ital. biol. sperim., 1958, v. 34, p. 1397. 115. Friend D., Zileli S., Hamlin J., Reuller F. — J. Clin, exper. Psychopathol., 1958, v. 19, Suppl 1, p. 61. 116. Gaddum J. — J. Physiol., 1953, v. 121, p. 15P. 117. Gaddum J., Giarman N. — Brit. J. Pharmacol., 1956, v. 11, p. 88 118. Gaddum J., Hameed K. — Brit. J. Pharmacol., 1954, v. 9, p. 240. 119. Gaddum J., Hebb C, Silver A., Swan A. — Quart J. exper. Physiol., 1953, v. 38, p. 255. 120. Gahen R., Nadaud J., Aubron S. — Therapie, 1961, v. 16, p. 322. 121. Gal E., Drewes P. — Nature, 1961, v. 189, p. 234. 122. Gal E., Drewes P., Barraclough C. — Proc. 1st internat. Pharmacol. Meet. v. 8, Oxford — London — New York — Paris, 1961, p. 107. 123. Gallagher W., Schroeppel A., Pfeiffer С — Arch. Gen. Psychiat., 1959, v. 1, p. 215. 124. Garattini S. — Schweiz. Arch. Neurol. Psychiat., 1959, v. 84, p. 269. 125. Garattini S., Katо R., Valzelli L. — Psychiat. Neurol., 1960, Bd. 140, p. 190. 126. Garattini S., Valzelli L. — Boll. Soc. ital. biol. sperim., 1955, v. 31, p. 1648. 127. Garattini S., Valzelli L. — Boll. Soc. ital. biol. sperim., 1956, v. 32, p. 288. 128. Garattini S., Valzelli L, — Boll. Soc. ital. biol. sperim., 1956, v. 32, p. 292. 129. Garattini S., Valzelli L. — Science, 1958, v. 128, p. 1278. 130. Garello L., Dolce G. — Sistema. Nerv., 1956. v. 8, p. 61. 131. Garven J. — Brit. J. Pharmacol., 1956, v. 11, p. 66. 132. Geiger R. — Fed. Proc., 1958, v. 17, p. 52. 133. Geller W. — Psychiat. Neurol., 1960, Bd. 140, S. 55. 134. Gennes L., de Fossey M. — Presse med., 1956, v. 64, p. 1066. 135. Gey K., Pletscher A. — J. Pharmacol, exper. Therap., 1961, v. 133. p. 18. 136. Giarman N. — Fed. Proc., 1961, v. 20, p. 897. 137. Giertz H. — Dtsch. Med. J., 1961, Bd. 12, S. 303. 138. Glasson B. — Rev. med. Suisse romane, 1958, v. 78, p. 419. 139. Glow P. — J. Neurol. Neurosurg. Psychiat., 1959, v. 22, p. 11. 140. Gluckman M., Hart E., Marrazzi A. — Science, 1957, v 126, p. 448. 141. Goldman D. — Ann. N. Y. Acad. Sci., 1959, v. 80, p. 687. 142. Gomirato G., Zanalda A. — Arch. ski. med., 1958, v. 106, p. 323. 143. Gordon P., Haddy F., Lipton M. — Science, 1958, v. 128, p. 531. 144. Grana E., Lilla L. — Farmaco. sci., 1957, v. 12, p. 1025. 145. Grandjean E., Battig K. — Helv. physiol. Pharmacol. acta, 1957, Bd. 15, S. 366. 146. Granowitz E., Pletscher A. — Helvet. med. acta, 1957, v. 24 p. 21. 147. Green J., Paasonen M., Giarman J. — Proc. Soc. exper. Biol. Med., 1957, v. 94, p. 428. 148. Greig M., Gibbons A. — Arch. internat. pharmacodyn., 1962, v. 136. p. 147. 149. Gursey D., Olson R. — Proc. Soc. exp. Biol. Med., 1960, v. 104, p. 280. 150. Gyermek L. — Lancet, 1955, p. II, p. 724. 151. Gyermek L., Gergely Y., Csak Z. — Acta pharm. Hung., 1957, v. 27, p. 66. 152. Gyermek L., Lazar I., Csok A. — Arch. internat. pharmacodyn., 1956, v. 107, p. 62. 153. Hart E., Соhn V., Marazzi A. — J. Pharmacol, exper. Therap., 1958, v. 122, p. 30A. 154. Haverback В., Dutсher T., coll. — N. Engl. J. Med., 1957, v. 256. p. 343. 155. Haverback В., Sjoerdsma A., Terry L. — N. Engl. J. Med., 1956, v. 255, p. 270. 156. Hehman K., Vonderahe A., Peters J. — Neurology, 1961, v. 11, p. 1011. 157. Hess S., Shоre P., Brodie В. — J. Pharmacol, exper. Therap., 1956, v. 118, p. 84. 158. Himwich H. — J. Nerv. Ment. Dis., 1955, v. 122, p. 413. 159. Himwich H. — Dis. Nerv. Syst., 1956, v. 17, p. 109. 160. Himwich H. — Rec. Advan. biol. Psychiat., 1961, v. 3, p. 77. 161. Himwich H. — Amer. J. Psychiat., 1959, v. 115, p. 756. 162. Hoagland H. — Ann. N. Y. Acad. Sci., 1957, v. 66, p. 445. 163. Hoagland H. — J. Nerv. Ment. Dis., 1958, v. 126, p. 211. 164. Holtz P. — Dtsch. med. Wschr., 1958, v. 16, S. 681. 165. Holzbauer M., Vogt M. — J. Neurochem., 1956, v. 1, p. 8. 166. Hsia D., Rowley W., Huang I. — J. Ment. Defic. Res., 1961, v. 5, p. 4. 167. Hurst L. — Med. Proc. (S. Africa), 1961, v. 7, p. 417. 168. Jоgues R., Вein H., Meier R. — Helv. physiol. pharmac. Acta, 1956, v. 14, p. 269. 169. Jatzkewitz H. — Confinia neurol., 1958, Bd. 18, S. 96. 170. Jensen E., Kodahl Т., Reiter P. — Acta psych, neurol. Scand., 1959, v. 34, Suppl. 136, p. 383. 171. Jillet R. — J. Ment. Sci., 1960, v. 106, p. 699. 172. Jus A., Laskowska D., Zimny S. — Ann. med. psychol., 1958, v. 116, p. 898. 173. Jus A., Laskowska D., Zimny S. — Neurol. Neurochir. Psychiat. pol., 1959, v. 9, p. 233. 174. Jus A., Laskowska D., Zimny S. — Psychiat. Neurol, med. Psychol., 1960, Jg. 12. p. 241. 175. Jus A., Laskowska D., Zimny S. — Neurol. Neurochir. Psychiat. polska, 1961, v. 11, p. 353. 176. Kamijо К., Koelle G., Wagner H — J. Pharmacol, exper. Therap., 1956, v. 117, p. 213. 177. Kaneko V., MсСubbin J., Page I. — Circulat. Res., 1960. v. 8, p. 1228. 178. Karki N., Paasonen M. — Acta pharmacol. toxicol., 1959, v. 16, p. 20. 179. Karki N., Paasonen M. — J. Neurochem., 1953, v. 3, p. 352. 180. Kety S. — Science, 1959, v. 129, p. 1528. 181. Kety S. — Science, 1959, v. 129, p. 1590. 182. Kety S. — Fed. Proc. 1961, v. 20, p. 894. 183. Kimball R., Friedman A., Vallejo E. — Neurology. 1960. v. 10, p. 107. 184. Kimura E., Young P., Richards R. — J. Allergy, 1960, v. 31, p. 237. 185. Kind H., Schneider H. — Dtsch. med. Wschr., 1957, Bd. 82, S. 1731. 186. Kirshner N., Goodall Ms. C., Rosen L. — J. Pharmacol. exper Therap., 1959, v. 127, p. 1. 187. Kivalo E., Rinne U., Karinkanta H. — J. Neurochem., 1961, v. 8, p. 105. 188. Klaus D., Wenzel E. — Z. ges. exper. Med., 1958, Bd. 130. S. 467. 189. Klee G., Bertino J., Goodman A., Aronson H. — J. Ment Sci., 1960, v. 106, p. 309. 190. Knoll J., Knoll B. — Arch. internat. pharmacodyn.. 1961, v. 133, p. 310. 191. Kobinger W. — Acta pharmacol. et toxicol., 1958, v. 14, p. 138. 192. Kobinger W, — Arch. f. exper. Pathol. Pharmakol., 1958. Bd. 233, p. 559. 193. Koch P., Laurin C, Lefebvre P., Bourdon P. — Amer. J. Psychiat., 1961, v. 118, p. 457. 194. Kuntzman R., Mead J., Brodie B., Shore P. — J. Pharmacol. exper. Therap., 1958, v. 122, p. 41. 195. Kveder S., Mclsaac W. — J. Biol. Chem., 1961, v. 236, p. 3214. 196. Labоrit H., Соirault R., coll. — Ann. med. Psychol., 1958. v. 2, p. 60. 197. Lafоn M., Воуer, coll. — Ann. med. Psychol., 1956, v. 114, p. 463. 198. Lafоn R., Duс N.. coll. — Therapie, 1957, v. 12, p. 371. 199. Lauer J. — J. clin. exper. Psychopathol., 1958, v. 19, Suppl 1, p. 110. 200. Lauer J., Inskip W., Bernsohn J., Zeller A. — Arch. Neurol. Psychiat., 1958, v. 80, p. 122. 201. Leoni G., Patnelli A. — Neuropsichiatria, 1955, v. 21, p. 37. 202. Lerner A., Case J., Takahashi Y. — J. Biol. Chem., 1960. v. 235, p. 1992. 203. Lessin A., Parkes M. — J. Pharmacy a. Pharmacol., 1957, v. 9, p. 657. 204. Levу J., Miсhel-Ber E. — Encephale, 1962, v. 51, p. 132. 205. Ljungberg E. — Schweiz. Arch. Neurol. Psychiat., 1961, Bd. 87, S. 351. 206. Mantegazzini P. — Boll. soc. ital. biol. sperim., 1956, v. 32, p. 839. 207. Mantegazzini P. — Arch. internat. pharmacodyn., 1957. v. 112, p. 199. 208. Mariani E., Rede G., Mоntanari F. — Riv. Neurol., 1960, v. 30, p. 195. 209. Marrazzi A. — Ann. N. Y. Acad. Sci., 1957, v. 66, p. 496. 210. Marrazzi A. — Amer. J. Psychiat., 1960, v. 116, p. 911. 211. Marrazzi A. — Ann. N. Y. Acad. Sci, 1962, v. 96, p. 211. 212. Marrazzi A., Hart E. — Science, 1955, v. 121, p. 365. 213. Marrazzi A., Hart E. — J. Nerv. Ment. Dis., 1955. v. 122, p. 453. 214. Marrazzi A., Hart E. — J. Nerv. Ment. Dis., 1956, v. 124. p. 388. 215. Marrazzi A., Hart E., Соhn V. — Progr. Neurol. Psychiat., 1956 Jr. 11, p. 565. 216. Marchall E., Stirling G., Tait A., Tоdrick A. — Brit. J. Pharmacol., 1960, v. 15, p. 35. 217. Martin G., Eriksen N., Вenditt E. — Fed. Proc., 1958, v. 17, p. 447. 218. Masuda M., Slonecker S., Doprat J. — J. Nerv. Ment. Dis., 1960, v. 130, p. 125. 219. McCubbin J, Kaneko V., Page I. — Circulat. Res., 1960, v. 8 p. 849. 220. Mead J., Shore P., Weissbach H., Leeper L., Brodie B. — J. Pharmacol. exper. Therap., 1958, v. 122, p. 51. 221. Miline R., Stern P., Hukovic S. — Experientia, 1958, v. 14, p. 415. 222. Mоnnier M., Tissоt R. — Schweiz. Arch. Neurol. Psychiat., 1958, v. 22, p. 218. 223. Mоnnier M., Tissоt R. — Helv. physiol. Pharmacol. acta, 1958, v. 16, p. 255. 224. Moreno V. — Acta Neuropsiquiat. Argent., 1961, v. 7, p. 216. 225. Nakazawa T. — Folia psychiatr. neurol. jap., 1960, Suppl. № 6. p. 12. 226. Olsоn R., Gurseу D., Vester J. — N. Engl. J. Med., 1960, v. 263 p. 1169. 227. Ozaydin S., Turker K., Koptagel G. — Neurol. Psychiat., 1961, v. 14, p. 967. 228. Paasonen K., Giarman N. — Arch. intern. pharmacodyn., 1958, v. 114, p. 189. 229. Paasonen M., Vogt M. — J. Physiol., 1956, v. 131, p. 617. 230. Page I. — Ann. N. Y. Acad. Sci., 1957, v. 66, p. 592. 231. Page I. — Science, 1957, v. 125, p. 721. 232. Page I. — Physiol. Rev., 1958, v. 38, p. 277. 233. Pare C., Sandler M., Stacey R. — Lancet, 1957, v. 1, p. 551. 234. Pare C., Sandler M., Stacey R. — Arch. Dis. Childr., 1959, v. 34, p. 422. 235. Pare C., Sandler M., Stacey R. — J. Neurol. Neurosurg. Psychiat., 1960, v. 23, p. 341. 236. Peart W., Andrews Т., Robertson J. — Lancet, 1961, p. I, p. 577. 237. Peсile A., Valseссhi A. — Boll. soc. ital. biol. sperim., 1959, v. 35, p. 726. 238. Pennes H. — Bull. N. Y. Acad, med., 1957, v. 33, p. 81. 239. Petrantoni S., Perris C, Bonetti U. — Cervello, 1959, v. 35, p. 97. 240. Pierre R., Сahn J. — C. r. Soc. biol., 1955. v. 49, p. 1406. 241. Platania S., Catanzaro A. — Acta Neurol., 1957, v. 12, p. 158. 242. Pletscher A. — Schweiz. med. Wschr., 1957, S. 1532. 243. Pletscher A. — Science, 1957, v. 126, p. 507. 244. Pletscher A., Besendorf H., Вachtold H. — Confinia neurol., 1958, Bd. 18, S. 137. 245. Pletscher A., Gey K. — Psychiat. Neurol., 1960, Bd. 140, S. 165. 246. Pletscher A., Shore P. — J. Pharmacol, exper. Therap., 1956, v. 116, p. 9. 247. Pletscher A., Shore P., Brodie B. — Science, 1955, v. 122, p. 374. 248. Pletscher A., Shore P., Brodie B. — J. Pharmacol. exper. Therap., 1956, v. 116, p. 46. 249. Pletscher A., Shore P., Brodie B. — J. Pharmacol. exper. Therap., 1956, v. 116, p. 84. 250. Poloni A. — Cervello, 1955, v. 31, p. 355. 251. Poloni A. — Cervello, 1956, v. 32, p. 123. 252. Poloni A. — Cervello, 1956, v. 32, p. 243. 253. Pscheidt G., Issekutz B.. Himwich H. — Quart. J. Stud. Alc., 1961, v. 22, p. 550. 254. Puca A., Lampitella P. — Acta Neurol., 1960, v. 15, p. 207. 255. Quivi D. — Le Sang, 1959, v. 30, p. 903. 256. Robins E., Lowe I., Havner N. — Clin. Res. Proc. 1956, v. 4, p. 149. 257. Rodnight R., McIlwain H. — Brit. Med. J., 1956, p. I, p. 108. 258. Roos B., Werdinius B. — Life Sci., 1962, № 3, p. 105. 259. Rudу L., Соsta E., Rinaldi F., Himwiсh H. — J. Nerv. Ment. Dis., 1958, v. 126, p. 284. 260. Rysanek K., Vitek V. — Experientia, 1959, v. 15, p. 217. 261. Sacchi U., Вrusa A., Sоriani S. — Sistema Nerv., 1957, v. 9, p. 230. 262. Sacchi U., Garello L., Dolce G., Bonamini F. — Boll. soc. ital. biol. sperm., 1955, v. 31, p. 663. 263. Sacchi U., Gianniotti G. — Boll. Soc. ital. biol. sperim. 1956, v. 32, p. 175. 264. Sano I. — Confinia neurol., 1958, v. 18, p. 196. 265. Sano I., Kakimoto Y., Okamoto T. — Nervenarzt, 1957, Jg. 28, S. 363. 266. Sano I., Kakimoto Y., coll. — Schweiz. Med. Wschr., 1957, Bd. 87, S. 214. 267. Santangelo R, Sinisi G. — Pisani, 1956, v. 70, p. 219. 268. Schain R., Freedman D. — J. Pediat., 1961, v. 58, p. 315. 269. Schanberg S., Giarman N. — Biochem. Pharmacol., 1962, v. 11, p. 187. 270. Schmid E., Kinzelmeier H., Seng I. — Experientia, 1959, v. 15, p. 230. 271. Schneider H. — Schweiz. Arch. Neurol. Psychiatr., 1958, Bd. 81, S. 344. 272. Schneider J., Sigg E., — В кн. Pennes H. Psychopharmacology, London, 1958, p. 75. 273. Sсhоu M. — Zbl. Neur., 1957, Bd. 139, S. 153. 274. Selbach H. — Psychiat. Neurol., 1960. Bd. 140, S. 69. 275. Shaw E., Wooley W. — Science, 1956, v. 124, p. 121. 276. Shore P., Brodie B. — Proc. Exper. Biol. Med., 1957, v. 94, p. 433. 277. Shore P., Brodie B. — Science, 1958, v. 127, p. 704. 278. Shore P., Pletscher A., Brodie B. — J. Pharmacol. exper. Therap., 1956, v. 116, p. 51. 279. Shore P., Pletscher A., coll. — Ann. N. Y. Acad. Sci., 1957, v. 66, p. 609. 280. Shore P., Pletscher A., coll. — J. Pharmacol, exper. Therap., 1956, v. 117, p. 232. 281. Shore P., Silver S., Brodie B. — Experientia, 1955, v. 11, p. 272. 282. Shore P., Silver S., Brodie B. — Science, 1955, v. 122, p. 284. 283. Sicuteri F. — Med. exptl., 1960. v. 2, p. 36. 284. Sicuteri F. — Clin, terap., 1961, v. 21, p 394. 285. Sicuteri F. — Boll. Soc. ital. biol. sperim., 1962, v. 38, p. 381. 286. Sicuteri F., Testi A., Anselmi B. — Int. Arch. Allergy, 1961, v. 18, p. 54. 287. Sjoerdsma A., Kornetsky C, Evarts E. — Arch. Neurol. Psychiat., 1956, v. 75, p. 488. 288. Sjoerdsma A., Oates J., Zaltzman P., Udenfriend S. — J. Pharmacol, exper. Therap., 1959, v. 126, p. 217. 289. Sjoerdsma A., Weissbach H., Undenfriend S. — Amer J Med., 1956, v. 20, p. 520. 290. Snow P., Lennard-Jones J., Curzon G., Stacey R. — Lancet, 1955, p. II, p. 1004. 291. Southren A., Warner R., Christoff N., Weiner H. — N. Engl. J. Med., 1959, v. 260, p. 1265. 292. Speсtоr S., Kuntzman R., Shore P., Brodie B. — J. Pharmacol, exper. Therap., 1960, v. 130, p. 256. 293. Speсtоr S., Prockop P., Shore P., Brodie B. — J. Pharmacol exper. Therap., 1958, v. 122, p. 71A. 294. Spies Т., Stоne R. — J. A. M. A., 1952, v. 150, p. 1599 295. Stumpf W. — Psychiat. Neurol., 1960, Bd. 140, S. 63. 296. Tabachnick I., Rubin A. — Proc. Soc. Exper. Biol. Med., 1959, v. 101, p. 435. 297. Tedeschi D., Tedeschi R., Fellows E. — Arch. internet, pharmacodyn., 1961, v. 132, p. 172. 298. Tedeschi D., Tedeschi R., Fellows E. — Rev. canad. biol., 1961, v. 20, p. 209. 299. Teruya Nagata M. E. — Arch. neurol. psiquiatr., 1959, v. 9, p. 118. 300. Thal N. — Dis. Nerv. Syst., 1959, v. 20, Sect. I, p. 197. 301. Tissоt R., Mоnnier M. — Helv. physiol. Pharmacol. acta, 1958, v. 16, p. 268. 302. Tоdrick A., Tait A., Marshall E. — J. Ment. Sci., 1960, v. 106, p. 884. 303. Tоnini G. — Boll. soc. ital. Biol, sperim., 1955, v. 31, p. 766. 304. Tоnini G. — Boll. soc. ital. Biol, sperim., 1958, v. 34, p. 793. 305. Truitt E. — J. Nerv. Ment. Dis., 1958, v. 126, p. 184. 306. Truitt E., Ebersberge E. — Science, 1962, v. 135, p. 105. 307. Twarog M., Page I. — Amer. J. Physiol., 1953, v. 175, p. 157. 308. Udenfriend S., Bogdanski D., Weissbach H. — Fed. Proc., 1956, v. 15, p. 493. 309. Udenfriend S., Titus E., Weissbach H. — J. biol. Chem., 1955, v. 216, p. 499. 310. Udenfriend S., Titus E., Weissbach H., Peterson R. — J. Biol. Chem., 1956, v. 219, p. 335. 310. Udenfriend S., Weissbach H., Bogdanski D. — Ann. N. Y Acad. Sci., 1957, v. 66, p. 602. 312. Udenfriend S., Weissbach H., Bogdanski D. — J. Biol. Chem., 1957, v. 224, p. 803. 313. Udenfriend S., Weissbach H., Sjordsma A. — Science, 1956, v. 123, p. 669. 314. Valcourt A. — Arch. Neurol. Psychiat., 1959, v. 81, p. 292. 315. Valdecasas F., Calvet F., Cuenca E., Salva J. — Arzneimittel-Forsch., 1956, Bd. S. 594. 316. Vassilia G., Costa E., coll. — Amer. J. Psychiat., 1961, v. 117, p. 1121. 317. Ventra D., Serra C. — Acta Neurol., 1956, p. 1112, p. 935. 318. Vincet D., Segonzac G. — Therapie, 1960, v. 15, p. 914. 319. Voelkel A. — Confin. Neurol., 1958, v. 18, p. 144. 320. Voelkel A. — Psychiat. Neurol., 1960, Bd. 140, S. 36. 321. Wada J. — Science, 1961, v. 134, p. 1688. 322. Walaszek E., Abood L. — Proc. Soc. exper. Biol. Med., 1959, v. 101, p. 37. 323. Warner R., Kirschner P., Warner G. — J. A. M. A., 1961, v. 178, p. 1175. 324. Weissbach H., Bogdanski D., coll. — J. Biol. Chem., 1957, v. 227, p. 617. 325. Weissbach H., Bogdanski D., Udenfriend S. — Arch. Biochem. Biophysics, 1958, v. 73, p. 492. 326. Weissbach H., Lovenberg W., coll. — J. Pharmacol. exper. Therap., 1961, v. 131, p. 26. 327. Winters W. — Marquette Med. Rev., 1959, v. 24, p. 204. 328. Woolley D. — Rend. Ist. super. sanita, 1956, v. 19. Suppl. p. 231 329. Woolley D. — Science, 1957, v. 125, p. 752. 330. Woolley D. — Res. Publ. Ass. Nerv. Ment. Dis., 1958, v. 36, p. 381, 331. Woolley D. — В кн. Pennes H., Psychopharmacology, London, 1958, p.152. 332. Woolley D. — Dis. Nerv. Syst., 1960, v. 21, Section II, p. 87. 333. Woolley D., Shaw E. — Science, 1954, v. 119, p. 587. 334. Woolley D., Shaw E. — Brit. Med. J., 1954, v. 2, p. 122. 335. Woolley D., Shaw E. — Ann. N. Y. Acad. Sci., 1957, v. 66, p. 649. 336. Yen C., Stanger R., Millman N. — Arch. internet, pharmacodyn., 1959, v. 123, p. 179. 337. Zaimis E. — Progr. med., 1958, v. 86, p. 411. 338. Zanowiak P., Rodman M. — J. Amer. Pharmacol. Assoc., Scient. Ed., 1959, v. 48, p. 165. 339. Zeller E. — J. clin. exper. Psychopathol., 1958, v. 19, Suppl. I, p. 106. 340. Zeller E., Bernsohn J., Inskip W., Lauer J. — Naturwissenschaften, 1957, v. 44, p. 427. 341. Zettler G., Sсhlоsser L. — Arch. exper. Path. u. Pharmak., 1954, Bd. 222, S. 345. 342. Zuanazzi G., Rognoni F. — Atti. Soc. Lombarda sci. med. biol., 1959, v. 14, p. 305. Глава 2 ОБМЕН АДРЕНАЛИНА И ПСИХИЧЕСКИЕ НАРУШЕНИЯ Адреналин и его роль как медиатора симпатической нервной системы известны уже с начала нынешнего столетия. Вопросу о синтезе и расщеплении адреналина посвящено огромное количество работ. Изложение этих данных приведено в соответствующих руководствах и не является нашей задачей. Мы лишь кратко приведем те основные выводы, которые имеют значение для оценки гипотез, касающихся роли адреналина в возникновении психических заболеваний. Адреналин и норадреналин синтезируются из более простых ароматических аминов и аминокислот, вероятно, из фенилаланина. Непосредственными предшественниками адреналина являются диоксифенилаланин (ДОФА) и образующийся из него окситирамин (более известный под названием допамин) который, окисляясь, переходит в норадреналин и посредством метилирования — в адреналин (фиг. 1). 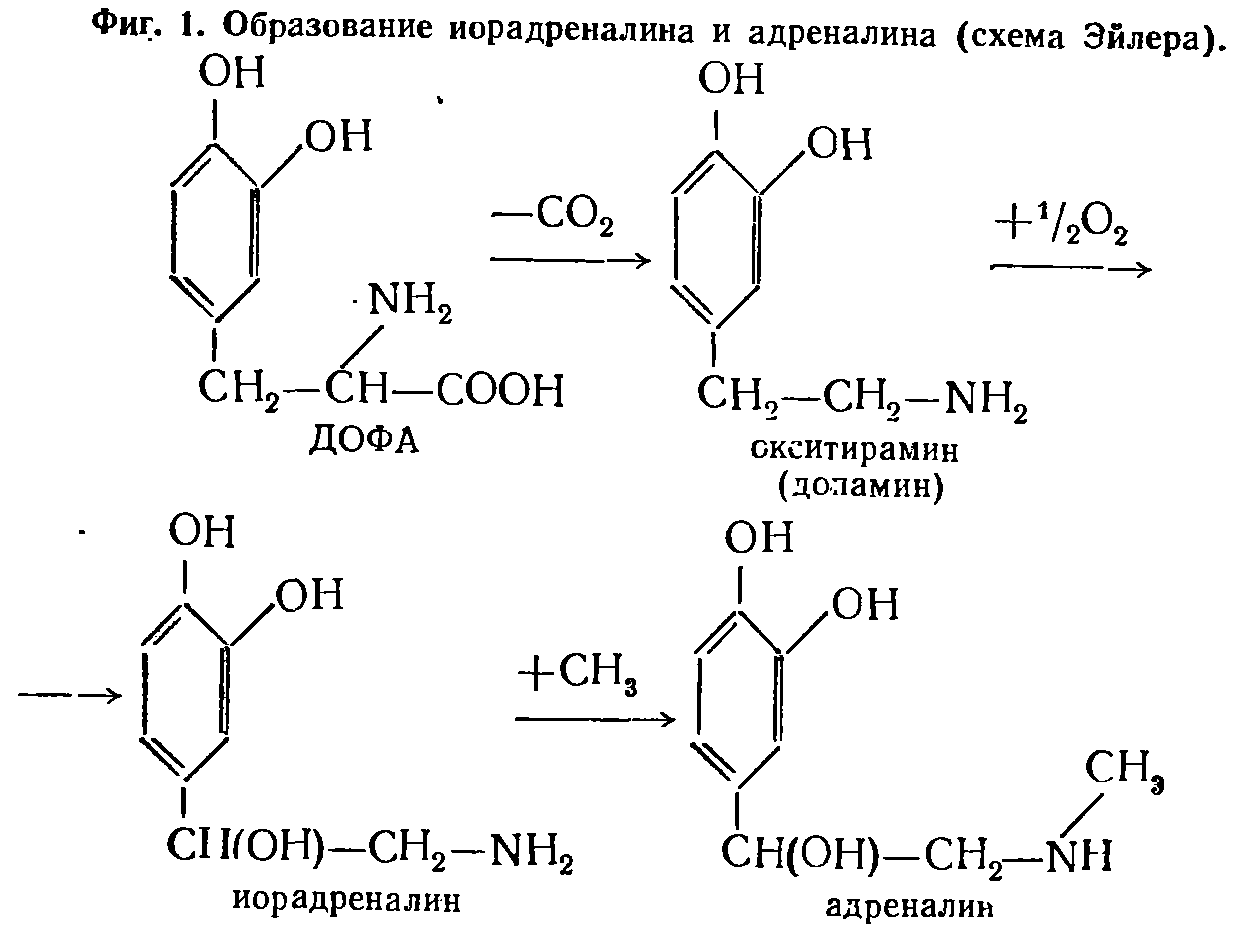 В головном мозгу норадреналин обнаружен в 1946 году фон Эйлером и в 1950 году Гольцем. В экспериментах на животных было показано, что норадреналин в различных отделах головного мозга распределяется неравномерно. Наиболее высокая его концентрация обнаружена в гипоталамической области. Относительно высокие концентрации норадреналина обнаружены также в сером веществе вокруг Сильвиева водопровода, в ряде других структур среднего мозга, обонятельном мозге, несколько меньшие — в ретикулярной формации ствола головного мозга, на дне 4-го желудочка в медиальных ядрах таламуса. В остальных структурах головного мозга норадреналин обнаружен лишь в виде следов. Допамин обнаружен в покрышке и в хвостатом ядре, где он, по-видимому, играет физиологическую роль, в остальных участках мозга он, очевидно, быстро превращается в норадреналин и концентрация его незначительна. С возрастом у животных возрастает как абсолютная, так и относительная (в расчете на 1 г вещества) концентрация норадреналина в головном мозгу. Норадреналин считают основным медиатором симпатической нервной системы в головном мозгу, где его концентрация по разным данным в 3—10 раз превосходит концентрацию адреналина. Однако имеются указания, что адреналин действует на нервную систему в несколько раз сильнее, чем норадреналин (при равных дозах). В мозгу не найдены ферменты, участвующие в образовании ДОФА, как и 5-гидрокситриптофана (5-ГТФ), но обнаружены декарбоксилазы, превращающие ДОФА в норадреналин (и 5-ГТФ в серотонин), как упоминалось в разделе, посвященном серотонину, ДОФА и ГТФ-декарбоксилазы идентичны. Очевидно, ДОФА и 5-ГТФ образуются вне мозга, а затем, проникая через гемато-энцефалический барьер, превращаются в серотонин и норадреналин, которые фиксируются тканями и фармакологически неактивны. Существование связанной и свободной формы не доказано, но весьма вероятно. Согласно схеме, предложенной Коста и сотр., оба основных моноамина — серотонин и норадреналин — содержатся в нервной клетке в виде скоплений, резервного и мобильного, между которыми поддерживается постоянное равновесие. Эти скопления отделены липоидной мембраной от части клетки, в которой находится инактиватор моноаминов — МАО, а также рецепторы, воздействуя на которые они осуществляют свою основную функцию — повышение симпатического (норадреналин) или парасимпатического (серотонин) тонуса. Под влиянием нервного импульса моноамины проникают через мембрану, воздействуют на рецепторы и разрушаются МАО, проникновение через мембрану осуществляется также за счет процесса диффузии, подчиняющейся физико-химическим законам. Согласно этой схеме активную роль играют только свободные моноамины. Различные гипотезы, посвященные роли норадреналина и адреналина при психических заболеваниях учитывают только их роль как медиаторов симпатической нервной системы, хотя катехоламины оказывают влияние на ряд обменных процессов, происходящих в организме, в частности на углеводный обмен нервной клетки. Адреналин по своему строению напоминает индол с незамкнутым кольцом и по своему строению близок к психотомиметическому средству — мескалину. На фиг. 2 приведены формулы адреналина и мескалина и двух метаболитов адреналина — адренохрома и адренолютина. Этим двум метаболитам придается наибольшее значение в генезе психических нарушений.  В организме адреналин быстро расщепляется. В отечественных руководствах выделено 3 основных пути обмена адреналина: 1) Окислительное дезаминирование под влиянием моноаминоксидазы с образованием 3, 4-диоксиванилиновой кислоты. 2) Образование ортохинона, превращающегося в адренохром и лейкоадренохром и, в конечном итоге, — в меланин. 3) Образование эфиров серной кислоты. Большинство зарубежных авторов выделяет 2 основных пути метаболизма адреналина: 1) Окислительное дезаминирование и 2) О-метилирование с образованием метанефрина. Как видно из схемы обмена адреналина, обычно в обоих процессах участвуют оба основных фермента — МАО и метилтрансфераза, действие которых обнаруживается в каждом случае на разных этапах метаболизма адреналина (или норадреналина) (см. фиг. 3). Однако считают, что в ЦНС основную роль играет МАО, а метилтрансфераза участвует, главным образом, в обмене катехоламинов на периферии. 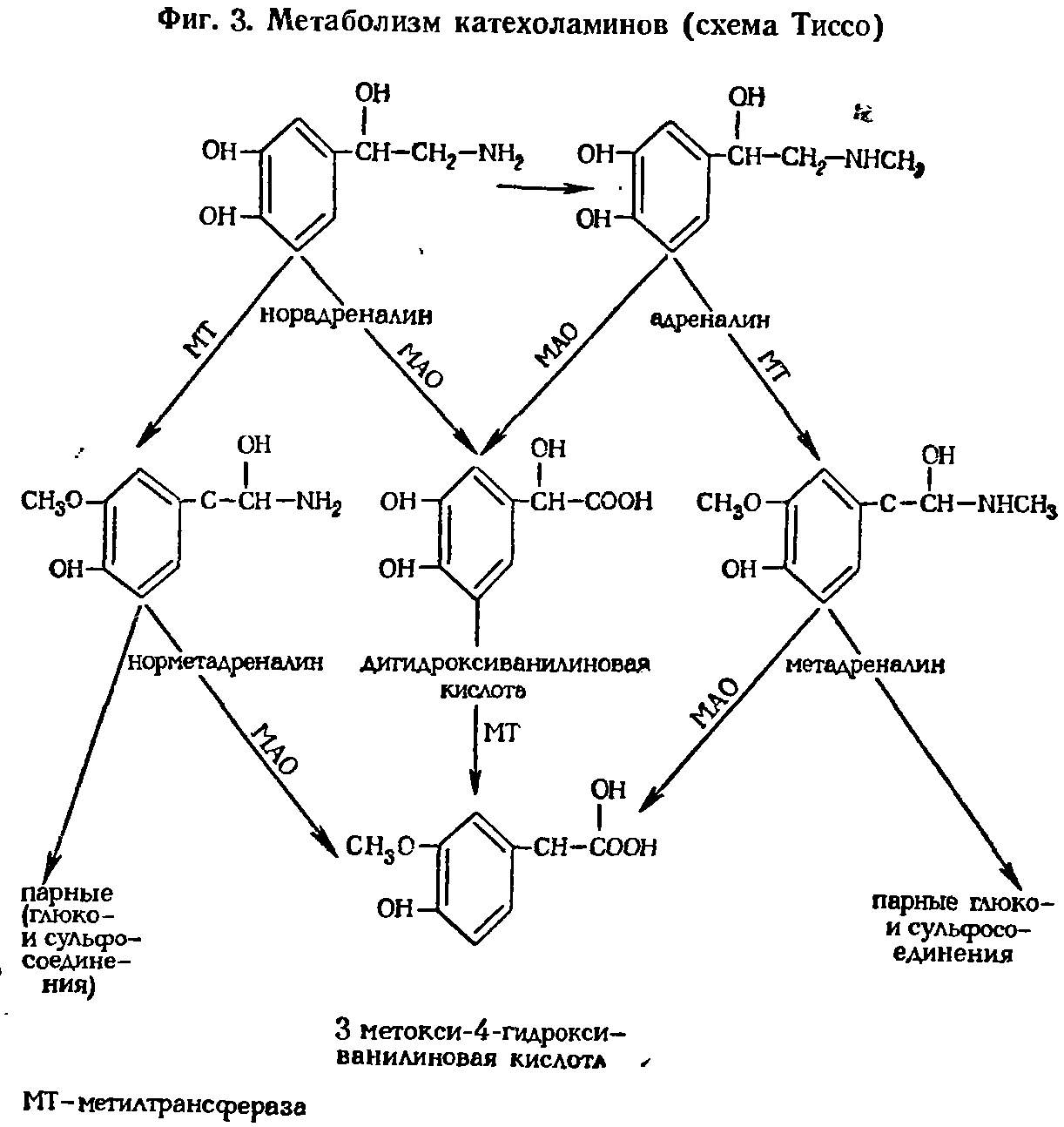 Есть все основания полагать, что норадреналин является медиатором эрготропной системы (по Гессу — центральная (подкорковая) система, координирующая вегетативные, соматические и психические функции, она подразделяется подобно периферической вегетативной нервной системе на эрготропную (симпатическую), подготавливающую организм к активной деятельности, и трофотропную (парасимпатическую), осуществляющую восстановительные процессы. Возможно, что выделенная после исследований Мэгуна и Моруцци восходящая, активизирующая ретикулярная система идентична эрготропной системе Гесса или является ее составной частью). В пользу этого свидетельствует распределение норадреналина и участвующих в его синтезе и расщеплении энзимов в головном мозгу. Мы не касаемся подробно вопроса об изменениях в содержании адреналина, развивающихся во время эмоциональных реакций у психически здоровых людей. Они впервые были описаны Кэнноном и подтверждены рядом последующих работ. Отметим лишь, что разнообразие эмоций, особенно человеческих, с трудом могло быть объяснено только увеличением концентрации в крови адреналина. В последние годы получены данные, показывающие, что эмоции сопровождаются изменениями содержания не только адреналина, но и близких к нему по строению веществ и что от характера этих изменений зависит и характер эмоциональной реакции. Так, исследование психически здоровых людей (спортсменов, студентов) и психически больных, показали, что при состоянии страха надпочечники выделяют большее количество адреналина, а при состояниях гнева или ярости увеличивается главным образом, количество норадреналина. У животных также во время бегства или защиты увеличивается выделение адреналина, а при нападении — норадреналина. По данным Гарба изупрель, амин, неотличимый от изопропил-норадреналина, в норме содержащийся в надпочечниках в очень малых количествах, вызывает чувство тревоги, являясь «медиатором чувства тревоги», при комбинированном действии адреналина и изупреля возникает подавленное настроение. Однако изучение связи обмена адреналина с психическими заболеваниями связано, главным образом, с 2-мя вопросами: 1) с гипотезой Осмонда, Смизис и Хоффера о нарушениях обмена адреналина, как причине шизофрении; 2) с изучением обмена моноаминов при депрессивных состояниях. В 1951 году Осмонд и Смизис, заметив сходство химической формулы адреналина и хорошо известного психотомиметического средства — мескалина (см. фиг. 2) предположили, что в определенных — патологических — условиях адреналин может превращаться в организме в вещество, условно названное ими М-веществом, сходное по своему действию с мескалином, и что это вещество является фактором, ведущим к развитию шизофрении. В дальнейшем эта гипотеза была поддержана Хоффером, который вместе с рядом своих сотрудников и по настоящее время активно ее разрабатывает. Правомерность такого предположения подтверждали отдельные наблюдения, когда адреналин вызывал психические нарушения. Так, д-р Асквит сообщил, что во время 2-ой мировой войны английские военные врачи пытались применить «розовый» (т. е. частично разложившийся) адреналин, но вынуждены были отказаться от этих попыток, т. к. при этом нередко у больных развивались психические нарушения. Один известный английский историк, которому Смизис излагал свою гипотезу, сообщил автору, что у него адреналин, который он, страдая бронхиальной астмой, принимал в течение длительного времени, усиливал галлюцинации, отмечавшиеся у него с детского возраста. Впоследствии Хоффер, Осмонд и Смизис сообщили еще о 4 больных бронхиальной астмой, у которых прием адреналина вызывал кратковременные психические нарушения. Один из этих больных отметил, что он становился в это время равнодушным к людям, так, ведя по улице автомобиль, он совершенно не беспокоился, что под машину могут попасть дети. Другой больной после приема ДЛК вспомнил, что такое же состояние у него вызывал прием адреналина. В 1959 году Осмонд и Хоффер подробно описали историю болезни некоего м-ра Ковиша, который около 4-х недель подряд принимал адреналин из-за приступов бронхиальной астмы. В период приема адреналина у больного развились нарушения восприятия, мышления и эмоций, весьма сходные с наблюдаемыми при шизофрении. Эти изменения исчезли после прекращения приема адреналина. Сам больной, прочитав впоследствии описание психических нарушений, вызываемых у психически здоровых добровольцев адренохромом, нашел, что эти нарушения весьма сходны с изменениями психики, которые были у него. Смизис приводит личное сообщение Химвича, наблюдавшего параноид после введения 1 мг адреналина. Одна из наблюдавшихся нами больных, также страдавшая бронхиальной астмой, сообщила, что у нее после введения адреналина в течение короткого времени в голове появлялись «странные мысли» и она переставала понимать смысл произносимых ею слов. Это сообщение в то время не привлекло к себе нашего внимания (больная поступила в больницу по поводу атропинного делирия, вызванного случайным приемом большой дозы t-rae Belladonnae) и лишь впоследствии — после ознакомления с работами Осмонда и Хоффера — мы обратили внимание на сходство реакции нашей больной с приведенными ими описаниями. Вполне вероятно, что в действительности подобные реакции на адреналин не так редки, как их описания в литературе. Таким образом, адреналин, или его производные, могут вызывать психические нарушения. Однако редкость подобных наблюдений, наряду с широким применением адреналина в клинике заставляет предположить, что изменения психики в этих случаях вызывал не сам адреналин, а какой-то образовавшийся из него токсический продукт. К косвенным данным, свидетельствующим в пользу предположения о роли адреналина в генезе психических нарушений, в частности шизофрении, относятся и наблюдения над психозами, вызываемыми симпатомиметическими веществами, которые в ряде случаев обнаруживают значительное сходство с шизофренией. Нередко, если факт злоупотребления симпатомиметиками (амфетамином, первитином) неизвестен врачу, больному ошибочно ставится диагноз шизофрении (см. гл. 6). Непосредственное изучение содержания адреналина и норадреналина в плазме крови и в моче больных шизофренией не дало определенных результатов. Большинство исследователей не нашло сколько-нибудь существенной разницы между концентрацией в плазме крови катехоламинов у больных шизофренией, по сравнению как с больными другими психическими заболеваниями, так и со здоровыми людьми. Только А. Л. Гамбург обнаружил у большинства исследованных им больных шизофренией более низкое содержание в крови адреналина, чем у здоровых людей и больных другими психическими заболеваниями, и известный, хотя и неполный, параллелизм между улучшением психического состояния и стойким повышением содержания в крови адреналина. В литературе имеются сообщения об изменениях содержания в крови адреналина при состояниях слабоумия — олигофрении, старческой деменции. Однако данные эти противоречивы, отмечено как снижение, так и повышение содержания адреналина в крови. Лич, Хис и сотр. отметили, что при добавлении к плазме крови больных шизофренией адреналина окисление его происходит быстрее, чем в плазме здоровых лиц. Эти данные не были подтверждены рядом исследователей, изучавших как скорость окисления адреналина и норадреналина при добавлении их к плазме в опытах in vitro, так и изменения концентрации этих веществ в крови в динамике после внутривенного введения адреналина и норадреналина. Сами Лич и Хис не считали ускорение окисления адреналина плазмой крови специфичным для больных шизофренией, т. к. с одной стороны, такое ускорение вызывает и плазма больных инфекционными заболеваниями и злокачественными новообразованиями, не обнаруживающих нарушений психики, а с другой, — скорость окисления адреналина плазмой у части больных шизофренией остается нормальной. Исследование выделения с мочой адреналина и норадреналина также не обнаружило существенных различий между больными шизофренией, психически здоровыми людьми и больными другими нервно-психическими заболеваниями. Правда, некоторые исследователи находили, что содержание катехоламинов в моче больных шизофренией выше, чем у здоровых людей, но такое же и более значительное увеличение было обнаружено у больных неврозами, при алкогольном галлюцинозе и белой горячке и у больных феохромоцитомой без нарушений психики. Более постоянным является усиленное выделение катехоламинов с мочой и повышение их содержания в крови при состояниях тревоги, напряженности, страха как сопровождающихся, так и не сопровождающихся двигательным возбуждением. Эти изменения, по данным Брюна и Пшайдта, появляются и исчезают одновременно с появлением и исчезновением эмоциональных реакций. В сущности, в этих случаях речь идет не об изменениях, характерных для психических заболеваний, а лишь о подтверждении отмеченной у здоровых людей связи между эмоциональными изменениями и выделением катехоламинов. По всей вероятности, усиленное выделение катехоламинов при неврозах, белой горячке, острой шизофрении является лишь отражением интенсивности их эмоциональных переживаний, равно как снижение концентрации катехоламинов у части больных шизофренией — следствием снижения эмоционального тонуса у этих больных. Отсутствие ясной корреляции между психическим заболеванием и содержанием в крови и моче адреналина и норадреналина делает практически недоказуемой неоднократно высказывавшуюся в литературе гипотезу о том, что благоприятный эффект инсулиношоковой терапии шизофрении обусловлен ее регулирующим влиянием на обмен катехоламинов. Изменения содержания в крови адреналина после введения инсулина описывались неоднократно, но нет достаточных доказательств, что эти изменения лежат в основе лечебного действия инсулина. Однако и отсутствие значительных изменений в содержании адреналина в крови не исключает возможности образования малых количеств токсических продуктов его обмена. Эксперименты с диэтиламидом лизергиновой кислоты (ДЛК) достаточно ясно показали, что минимальные количества токсического вещества, с трудом обнаруживаемые современными методами исследования, могут вызвать тяжелые психические нарушения. Хоффер и его сотрудники предположили, что таким токсическим продуктом, образующимся в процессе обмена адреналина, может быть адренохром или адренолютин. При этом до последнего времени основное внимание уделялось изучению обмена адренохрома. Сам Хоффер указывал, что для доказательства правомерности этого предположения нужны 3 ряда фактов: во-первых, адренохром и адренолютин должны обладать психотомиметическими свойствами; во-вторых, должно быть доказано их образование в организме; и в-третьих, обмен адреналина должен быть нарушен у больных шизофренией. Рассмотрим в предложенной Хоффером последовательности факты, добытые как сторонниками, так и противниками этой гипотезы. а) Психотомиметическое действие адренохрома и адренолютина. Хоффер и ряд других исследователей нашли, что,адренохром вызывает нарушения поведения у различных экспериментальных животных — крыс, голубей, кошек. Введение адренохрома или адренолютина психически здоровым испытуемым вызывает у них нарушения восприятия, мышления и эмоциональности, сходные с изменениями, наблюдаемыми в начальной стадии шизофрении. Так, Хоффер, давая адренолютин 14 здоровым добровольцам, часто отмечал нарушения логического мышления — неспособность истолковывать пословицы, ошибки в счете и т. п. Объяснения испытуемых становились более конкретными и в то же время многословными, они часто приходили к решению окольными путями, нередко считали, что содержание предложенных им пословиц относится к ним лично. Тревога обычно уменьшается, сменяется чувством успокоения, иногда безразличия, утратой интереса к эксперименту, иногда отмечался неадэкватный смех. Исчезает критическое отношение к своему состоянию. В отдельных наблюдениях сходные изменения вызывала, однако, и дача плацебо, особенно при наличии чувства тревоги. По-видимому, тревога сама по себе ведет к увеличению содержания адреналина, а следовательно, — и адренолютина. Нарушения эти нередко весьма деликатно выражены и могут быть просмотрены, тем более, что адренохром и адренолютин уменьшают чувство тревоги и самочувствие испытуемых может даже улучшаться, несмотря на появление изменений мышления и восприятия. Незначительная выраженность психических нарушений по мнению Хоффера только подтверждает правильность его гипотезы: «Если есть химическое вещество у шизофреника, то оно должно вызывать незначительно выраженные изменения, иначе не было бы ни поздней диагностики, ни споров о том идет ли речь о шизофрении, депрессии, состояниях тревоги или о незрелости психики». В части случаев изменения психики были выражены значительно отчетливее. Приведем в качестве иллюстрации самонаблюдения Осмонда, вводившего себе до 10 мг адренохрома. В первый раз появилась необычная яркость красок и пластичность рисунка, при открытых глазах — цветные пятна, при закрытых глазах видел рыб, возникло ощущение, будто он лежит на дне моря. Затем появилось чувство, что в обычных вещах есть что-то неприятное, имеющее иной, скрытый смысл. Исчез интерес к опыту и его результатам, не хотелось разговаривать с окружающими. На следующий день отмечалась необычная яркость и чистота восприятия. Во втором эксперименте — когда доза адренохрома была увеличена, по сравнению с первым опытом, — более отчетливо было выражено равнодушие к происходящему и окружающим людям, от которых чувствовал себя отгороженным как будто стеклянным барьером; в то же время неодушевленные предметы вызывали к себе повышенный интерес. Появились неясные идеи отношения. Поведение Осмонда было необычным. Так, приведенный в общество, он ни с кем не поздоровался и в течение целого часа просидел молча, не принимая никакого участия в разговоре и рассматривая лежавший на полу коврик, — поведение в обычном состоянии ему не свойственное. Психозы, вызываемые адренохромом обнаруживают по данным Хоффера и Осмонда большее сходство с шизофренией, чем психозы, вызываемые мескалином и ДЛК. Интересны и самонаблюдения Хоффера и Осмонда над действием другого производного адреналина — адренолютина. У обоих после приема адренолютина на длительное время, исчисляемое неделями, исчезала способность эмоционально реагировать на происходящее. И тот и другой скрывали эти изменения друг от друга, опасаясь, что у них начинается шизофрения. Однако психотомиметические свойства адренохрома не были подтверждены другими исследователями, как в экспериментах на животных, так и в опытах на здоровых добровольцах. Правда, Таубман и Янц, давая 15 здоровым испытуемым 3 мг адренохрома, отметили легкие и преходящие вегетативные нарушения, изменения настроения — чаще в сторону подавленности. Менялся и цвет окружающих предметов, их пропорции, иногда испытуемым казалось, что предметы начинают двигаться. Однако все эти нарушения были кратковременными и исчезали уже спустя 30 мин. Длительных и отчетливых психических нарушений, описывавшихся Хоффером и Осмондом, вызвать не удалось. В другой серии экспериментов Хольц и Таубман не нашли изменений психики после дачи свежего адренохрома. Однако, когда они окисляли адренохром, встряхивая его на воздухе с сывороткой крови, и давали сублингвально депротеинизированную жидкость, у испытуемых развивался психоз. Ряд других исследователей, среди которых были и сторонники гипотезы Осмонда — Хоффера, не смогли повторить их опыты, несмотря на то, что в некоторых случаях доводили дозу до 50 мг чистого адренохрома. Более того, сами Хоффер и Осмонд не смогли повторить свои первые успешные эксперименты. Результаты проб, предпринимавшихся ими в течение ряда последующих лет, дали настолько неопределенные результаты, что у авторов, пользуясь их собственным выражением, возникла мысль, что быть может, адренолютин и адренохром не галлюциногенные средства, а только галлюцинация исследователей. Эти неудачи они впоследствии объясняли тем, что адренохром и адренолютин, использовавшиеся ими в эти годы, даже по своему внешнему виду отличались от тех же веществ, использовавшихся ими в первой серии экспериментов и полученных из других лабораторий, и что, по-видимому, разработанные в их лаборатории методы синтеза адренохрома и адренолютина были неточными. Только в конце 1957 — начале 1958 года удалось получить чистый адренохром и адренолютин. Однако, несмотря на то, что с этого времени прошло 5 лет, мы не смогли найти в литературе описаний опытов, подтверждающих первоначальные наблюдения авторов. Только в 1962 г. группа чешских исследователей сообщила об экспериментах на психически здоровых людях, у части которых развились после приема адренохрома психические нарушения, обнаруживавшие сходство с изменениями психики у больных шизофренией. При этом отмечалась значительная разница между действием адренохрома чехословацкого и зарубежного производства. Таким образом, способность адренохрома и адренолютина вызывать психические нарушения нуждается в дальнейшей проверке. Доза, по крайней мере, адренохрома, способная вызвать психические нарушения, относительно велика (по данным Хоффера и Осмонда — 10 мг, по данным Войтеховского и др. — 30 мг), если учесть, что содержание норадреналина в гипоталамусе — где он обнаруживается в максимальной концентрации — составляет всего около 0,001 мг на 1 г вещества мозга и что, по-видимому, только небольшая часть адреналина и норадреналина может превращаться в организме в токсические продукты. Более вероятно, что отмеченные при приеме адренохрома и адренолютина психические нарушения вызываются не самими этими веществами, а какими-то их производными, разное содержание которых и объясняет разнородность данных, полученных в перечисленных выше экспериментах. Впрочем, сами Осмонд и Хоффер не утверждали, что адренохром и адренолютин идентичны гипотетическому М-веществу, являющемуся, по их мнению, причиной или основным патогенетическим фактором шизофрении. Вопрос о том, содержится ли действительно адренохром в крови здоровых и психически больных — прежде всего, шизофренией, остается спорным. Положительные данные получены только Хоффером и его сотрудниками. Эта группа исследователей с помощью спектрофлюорографии обнаружила в крови как здоровых людей, так и больных различными психическими заболеваниями адренохром «или вещество, чрезвычайно близкое к нему». При этом общее содержание адренохрома в крови у здоровых, больных алкоголизмом, депрессивными состояниями и шизофренией оказалось одинаковым. Однако при шизофрении обнаружено нарушение распределения адренохрома между эритроцитами и плазмой. Соответствующий коэффициент составлял у депрессивных больных 3,4, у психически здоровых — 2,6, а у больных шизофренией — 1,7. Ремиссия сопровождалась нормализацией этого коэффициента. Таким образом, при шизофрении нарушена (по мнению Хоффера) способность эритроцитов адсорбировать адренохром. После внутривенного введения адренохрома содержание его в крови возрастает у больных шизофренией более значительно и увеличение это держится дольше, чем у здоровых и больных другими психическими заболеваниями. В спинномозговой жидкости больных шизофренией содержание адренохрома в 2 раза выше, чем в плазме, тогда как у не шизофреников (больных или психически здоровых) этот коэффициент равен единице. Что касается адренолютина, то прямых доказательств его наличия в крови, сколько нам известно, пока нет. Однако Хоффер и Кеньон, добавляя к крови здоровых людей адренолютин, отметили появление через 1 минуту вещества, аналогичного веществу, наблюдавшемуся ими в крови больных шизофренией через 80 минут после добавления к ней адреналина. Адренолютин в крови здоровых испытуемых на протяжении 80 минут не подвергался изменениям. Авторы приходят к выводу, что вещество, которое образуется сывороткой крови больных шизофренией и есть адренолютин. Лич и Хис также отметили на спектрофотограмме образование в крови через 80 минут после добавления к ней адреналина какого-то вещества, отличающегося от адренохрома. Вещество это образуется и у здоровых, поэтому нет оснований считать его специфически вызывающим психические нарушения. Однако содержание этого вещества в крови больных шизофренией было, по их данным, в среднем в 21/2 раза выше, чем у здоровых людей. Однако сама методика, которой пользовались Хоффер и его сотрудники, вызвала возражения со стороны ряда исследователей, которые не смогли подтвердить полученные Хоффером данные и пришли к выводу, что содержание в крови адренохрома как у здоровых, так и у больных шизофренией либо равно нулю, либо меньше той минимальной концентрации, которую позволяет определить как применявшийся группой Хоффера метод спектрофлюорографии, так и другие разработанные в настоящее время методы. Что же касается данных, полученных Хоффером, то по мнению Фелдстайна они являются артефактом: проверяя методику Хоффера он нашел, что свечение, интенсивность которого расценивалась как показатель концентрации в крови адренохрома, в действительности обусловлена реакцией уксуснокислого цинка и аскорбиновой кислоты. В развернувшейся полемике каждая из сторон доказывала правильность полученных ею данных. Не будучи специалистами в области спектрофлюорографии, мы не беремся судить о том, кто прав в этом вопросе. Однако, поскольку методика, на которой основаны цифры содержания адренохрома в крови и спинномозговой жидкости и скорость его окисления у здоровых и психически больных, подвергается сомнению, данные Хоффера и его сотрудников не могут считаться достоверными. Заметим, что спор касается только вопроса о содержании в крови адренохрома и что отсутствие — или минимальные количества — адренохрома в крови, хотя и лишает гипотезу о роли нарушений обмена адреналина в генезе шизофрении существенных аргументов, говорящих в ее пользу, недостаточно для того, чтобы ее отвергнуть. В последние годы Сулкович и сотр. сообщили о том, что ими обнаружен в моче здоровых и психически больных нестойкий фактор, подвергающийся окислению при хранении на холоду. Основываясь на том, что из известных метаболитов адреналина только адренолютин адсорбирует кислород из воздуха и что желтое свечение мочи, содержащей этот фактор аналогично свечению, получаемому при добавлении к моче адренохрома или адренолютина, авторы полагают, что нестойкий фактор содержащийся в моче — либо адренолютин, либо вещество, близкое к адренолютину. Поскольку сам адренолютин без добавления его к моче такого свечения не дает, более вероятным представляется второе предположение. Повышенное по сравнению с психически здоровыми людьми содержание в моче этого фактора обнаружено у большинства больных шизофренией, тогда как у больных соматическими заболеваниями без психических нарушений, больных неврозами, большинства больных маниакально-депрессивным психозом цифры были такими же, как у здоровых. У 2 больных шизофренией отмечено увеличение содержания этого фактора во время ухудшения состояния и уменьшение его при улучшении. Эти первые данные, свидетельствующие в пользу гипотезы о роли нарушений обмена катехоламинов, нуждаются в проверке, тем более, что связь обнаруженного нестойкого фактора с адренолютином еще не доказана. б) Действие адреналина на психически больных. В последние годы известный французский психиатр П. Абели и его сотрудники, основываясь на клинических наблюдениях, пришли к выводу, что в основе шизофрении лежит нарушение обмена и утилизации адреналина, по-видимому, вследствие поражения гипоталамической области. Абели отметил, что у значительной части больных дискордантным синдромом, особенно с большой давностью заболевания, отсутствуют брюшные рефлексы (как известно, Абели разделяет шизофрению на «дискордантный синдром», «шизоневрозы» и собственно раннее слабоумие, однако в дальнейшем мы для простоты изложения будем пользоваться термином шизофрения для обозначения этих больных, что нередко делает в своих работах и сам Абели). Исследуя действие различных лекарств, Абели и его сотрудники нашли, что только с помощью адреналина удается восстановить брюшные рефлексы. Одновременно улучшается и психическое состояние больных: исчезают — правда только на период дачи адреналина — расстройства восприятия, бредовые идеи, кататонический ступор и другие психопатологические симптомы, нормализуется ЭЭГ. Сходные данные о влиянии адреналина на больных шизофренией получил и О. И. Вольфовский, опубликовавший свои наблюдения примерно за 20 лет до появления работ Абели. Вводя группе больных шизофрении 1,5—2 мл адреналина ежедневно, он отметил постепенное побледнение продуктивных психопатологических симптомов, появление положительного отношения к лечению, у 11 из 36 больных с небольшой давностью заболевания, наступила ремиссия. Правда, у части больных становилась ярче эмоциональная окраска бредовых идей, которые становились более связными и больше коррелировали с поведением больных. Вряд ли такого рода динамика может расцениваться как улучшение. При хронической шизофрении положительных сдвигов в состоянии больных не было. Абели полагает, что шизофрения, может быть, обусловлена недостатком адреналина и что улучшение, наблюдаемое при инсулинотерапии, обусловлено вызываемой инсулином гиперсекрецией адреналина (механизм противорегуляции). Нетрудно заметить, что общим в гипотезах Хоффера и Абели является только признание роли нарушений обмена адреналина при шизофрении. Характер же этих нарушений противоположен. В то время как Хоффер считает, что при шизофрении образуется избыток адреналина или его метаболитов и задача врача — уменьшить секрецию адреналина, по мнению Абели, напротив, адреналина при шизофрении недостаточно и его следует вводить больным. Существенным подтверждением предположения о роли адреналина в генезе шизофрении, Абели считает опыты Минца Этот последний нашел, что введение кроликам сыворотки крови больных шизофренией обычно значительно уменьшает, устраняет, а иногда извращает прессорную реакцию, которая развивается при местном наложении адреналина на участок коры головного мозга кролика. Сыворотка здоровых людей, беременных женщин и больных, страдающих различными органическими заболеваниями головного мозга, такого действия не оказывает. У кроликов, получивших сыворотку крови больных шизофренией, концентрация адреналина и норадреналина в гипоталамусе значительно выше, чем у животных, которым вводилась сыворотка крови здоровых людей. Ипрониазид (ипразид), блокирующий расщепление адреналина и норадреналина моноаминоксидазой, подобно сыворотке крови больных шизофренией уменьшает или предотвращает подъем артериального давления, вызываемый местным наложением на кору головного мозга адреналина. Результаты опытов Минца свидетельствуют, таким образом, что сыворотка крови больных шизофренией ведет к повышению содержания катехоламинов в головном мозгу животных. Однако это не доказывает, что причиной этих изменений является недостаток адреналина в организме больных шизофренией. Предпринятая нами проверка наблюдений Абели и его сотрудников не смогла подтвердить полученных ими данных. Прежде всего, полное и двустороннее отсутствие брюшных рефлексов отмечено только у 5 из 50 обследованных больных, несмотря на то, что все они были больными с большой давностью заболевания, т. е. относились как раз к той категории больных, у которых, по данным Абели, выпадение брюшных рефлексов наблюдается наиболее часто. Во-вторых, ежедневное введение адреналина этим больным не нормализовало у них брюшных рефлексов и не улучшало их психического состояния. Напротив, у части больных наступило обострение заболевания, которое сгладилось после прекращения инъекций адреналина. Таким образом, основанная на предположении Абели попытка использовать адреналин как средство лечения больных шизофренией, дала результаты, скорее говорящие в пользу гипотезы Хоффера. Ухудшение состояния у наших больных после введения адреналина может быть истолковано как результат образования из него токсических метаболитов, т. е. говорит в пользу предположения о нарушении обмена адреналина при шизофрении, хотя непосредственное доказательство существования таких метаболитов пока, как мы упоминали, не получено (или, во всяком случае, недостоверно). Если правильна гипотеза Хоффера, то средства, нормализующие обмен адреналина, должны быть эффективным средством лечения шизофрении. Лечение может быть направлено на уменьшение общего количества адреналина, уменьшение превращения адреналина в адренохром и адренохрома — в адренолютин или способствовать выведению из организма либо нейтрализации этих веществ. Никотиновая кислота и никотинамид, являясь акцепторами метильной группы, уменьшают секрецию адреналина. Хоффер, Осмонд и их сотрудники получили хорошие результаты, применяя эти вещества при острой и хронической шизофрении, в частности, у некоторых больных шизофренией, прежде без успеха лечившихся инсулинотерапией или ЭКТ. «Ободряющие» результаты получены теми же авторами и при применении больших доз (10—20 г в день) аскорбиновой кислоты, которая, по их данным, препятствует превращению адреналина в адренохром, а адренохрома в адренолютин. Эти результаты не проверены другими исследователями. Нам известна только одна работа — Эшби и сотр., которые не получили никакого эффекта при лечении больных хронической шизофренией никотиновой кислотой и никотинамидом. Терапевтические попытки, основанные на гипотезе о роли адреналина, слишком немногочисленны, чтобы можно было придавать им существенное значение, но, несомненно, заслуживают дальнейшего изучения. Необходимо отметить, что в механизме действия аминазина (хлорпромазина), широко и с успехом применяющегося при лечении шизофрении, существенную роль, по-видимому, играет его адренолитическое действие, в частности, блокирование эрготропной системы. Этому вопросу посвящено огромное количество работ, суммировать которые здесь не представляется возможным и которые хорошо известны широкому кругу психиатров. Общая гипотеза Хоффера и Осмонда на протяжении последних лет подвергалась известным изменениям. С учетом последних данных она может быть сформулирована следующим образом. Основным патогенетическим звеном в патогенезе шизофрении является нарушение обмена адреналина с образованием токсических продуктов. Фактор, вызывающий эти нарушения, неизвестен. Этот гипотетический этиологический фактор у больных с определенной биохимической конституцией вызывает патологические изменения в вегетативной нервной системе (по признанию Хоффера и Осмонда — это наиболее слабое звено в их концепции). Начальной фазой этих изменений является усиление активности парасимпатической системы, затем присоединяется усиление активности симпатической системы (отчасти обусловленное холинергичностью симпатических ганглиев). В этой фазе и происходит образование токсических продуктов обмена адреналина. К таким токсическим продуктам относятся адренохром и адренолютин (более вероятно — последний), а может быть и другие, еще неизвестные вещества. Если в норме адренохром окисляется быстро и образует преимущественно лейкоадренохром, а за тем — дигидроксииндолы, то при шизофрении окисление адренохрома замедленно и идет с образованием преимущественно адренолютина с последующим превращением его в тригидроксииндолы. Несмотря на то, что основные звенья этой концепции на сегодняшний день остаются лишь подлежащими дальнейшей проверке гипотезой, с ее помощью пытаются объяснить некоторые клинические особенности больных шизофренией. Антимитотическим действием адренохрома объясняют пониженную сопротивляемость больных по отношению к туберкулезу, антиинсулинным — редкость у них диабета, антигистаминным — слабую выраженность аллергических реакций, которые, например, Ли обнаружил при шизофрении в 0,2% по сравнению с 3,4% у больных с последствиями черепномозговой травмы. Хоффер и его сотрудники попытались связать с нарушением обмена адреналина и такое распространенное заболевание, как алкоголизм. С их точки зрения одним из факторов, ведущих к алкоголизму, является состояние постоянной напряженности, обусловленное высоким коэффициентом адреналина : адренохрома. Адреналин увеличивает чувство напряженности, а адренохром (и адренолютин) его снижают. Алкоголь каким-то образом заменяет адренохром, который в организме больных алкоголизмом либо образуется в недостаточном количестве, либо исчезает быстрее, чем у здоровых. Способность алкоголя устранять напряжение приводит к постоянной потребности в приеме алкоголя. ДЛК, повышающий содержание в крови адренохрома и замедляющий его дальнейшее превращение, оказывает тем самым благоприятное действие на больных алкоголизмом. Все фактические данные, подтверждающие нарушения образования и метаболизма адренохрома при алкоголизме, получены с помощью той же методики спектрофлюорографии, о спорности которой упоминалось выше. Остановимся несколько подробнее на вопросе о связи между обменом адреналина и психотомиметическим эффектом ДЛК. В пользу этого предположения свидетельствуют эксперименты на кошках, в которых ДЛК усиливал сокращение мигательной перепонки, вызываемое адреналином. Однако такое же действие оказывали и производные ДЛК, лишенные психотомиметических свойств. В опытах на крысах адренохром не влиял на эффект ДЛК, а адренолютин оказывал на него тормозящее действие. Таким образом, в эксперименте не удалось показать потенцирующего действия адреналина и его метаболитов на эффект ДЛК. Хотя ДЛК вызывает симптомы как симпато-, так и парасимпатотонии, но превалирует симпатотоническое действие, что и дало основание полагать, что повышение тонуса симпатической (эрготропной) системы играет существенную роль в механизме психотомиметического действия ДЛК. В клиническом эксперименте было отмечено, что состояние «стрэсса», сопровождающееся повышением содержания в крови адреналина, облегчает возникновение ДЛК-психоза, который в этих случаях может быть вызван меньшими дозами ДЛК Напротив, состояние покоя, расслабленности затрудняет развитие психоза в ответ на прием ДЛК. Во время экспериментального психоза, вызванного ДЛК было также отмечено усиленное выделение с мочой адреналина и норадреналина и отсутствие этих изменений у тех больных, у которых ДЛК не вызвал психических изменений. Введение адренохрома или адренолютина после приема небольших доз ДЛК, которые сами по себе не вызывали психических нарушений, вело по наблюдениям Хоффера и Осмонда к развитию бурной психотической реакции, продолжавшейся около 6—9 часи характеризовавшейся расстройствами восприятия (изменения окраски предметов, искажение их формы и иллюзорное движение, неверная оценка расстояния, размеров, нарушения восприятия времени), мышления (остановка мыслей, конкретность мышления, бредовая настроенность), резкими сменами эмоций, неадэкватной активностью, негативизмом. Эта нарушения, по мнению авторов, характерны для адренохрома, а не для ДЛК, хотя мы не смогли уловить между ними существенного различия. Все эти наблюдения говорят в пользу предположения о роли симпатотонии в механизме развития, вызываемого ДЛК психоза, но не доказывают, что ДЛК действует на психику не непосредственно, а изменяя или усиливая обмен адреналина. Наиболее доказательным в этом отношении является усиленное выделение с мочой адреналина и норадреналина, но оно коррелирует не столько с психотомиметическим эффектом ДЛК, сколько с выраженностью периферических симпатотонических симптомов, которые могут быть устранены (например, с помощью дибензамина) без того, чтобы изменилась выраженность собственно психических нарушений. По аналогии с результатами опытов Минца можно трактовать как результат увеличения содержания в головном мозгу катехоламинов ослабление реакции на введение адреналина, отмечавшееся у лиц, получивших ДЛК, хотя аналогия с животными всегда остается лишь предположением. Точно так же и результаты паутинного теста — пауки, получившие ДЛК, ткут более тонкую паутину (паутина почти целиком состоит из адреналина) — доказывает только способность ДЛК влиять на обмен адреналина у паука, но вовсе не доказывает, что такое влияние имеет место в человеческом организме и что оно связано с психотомиметическим действием ДЛК. Наиболее демонстративные результаты, подтверждающие, связь психоза, вызываемого ДЛК, с обменом адреналина, получены группой Хоффера. Эти исследователи отметили увеличение содержания в крови адренохрома у психически здоровых людей и хронических алкоголиков, получивших ДЛК, замедление разрушения введенного внутривенно адренохрома, увеличение способности плазмы превращать адреналин в адренолютин. В то же время бром-ДЛК и морфолид лизергиновой кислоты, не вызывавшие психических нарушений, такого действия не оказывали. Эти данные могли бы служить важным доказательством связи психотомиметического эффекта ДЛК с нарушением обмена адреналина, если бы сама методика, с помощью которой были получены эти результаты, не подвергались сомнению, о чем мы упоминали выше. Следует отметить, что Хоффер и его сотрудники считают, что психоз вызываемый ДЛК, обнаруживает значительное сходство с острой шизофренией, хотя и не абсолютное, но, во всяком случае, более отчетливое, нежели сходство между свежезаболевшими и деградированными больными шизофренией. Поэтому, по их мнению, доказательство связи между действием ДЛК на психику и образованием патологических продуктов обмена катехоламинов явилось бы одновременно доказательством роли этих продуктов в генезе шизофрении. Сходство экспериментальных психозов с шизофренией оспаривается многими исследователями, уподобляющими ДЛК-психоз экзогенным типам реакции. Таким образом, роль нарушений обмена адреналина в генезе психических нарушений, возникающих после приема ДЛК, подтверждается рядом косвенных доказательств, представляется довольно правдоподобной, но прямых доказательств, подтверждающих правильность этого предположения, получить не удалось. Несмотря на то, что гипотеза о связи шизофрении с образованием продуктов обмена адреналина возникла в значительной степени благодаря сходству строения мескалина и адреналина, исследование взаимодействия мескалина и адреналина проводилось лишь в отдельных работах и не дало сколько-нибудь определенных результатов. В заключение кратко остановимся на вопросе о роли изменений обмена адреналина при маниакально-депрессивном психозе и при депрессивных состояниях другого происхождения. Интерес к этому вопросу в значительной степени связан с изучением механизма действия новых антидепрессивных средств — ингибиторов МАО и тофранила (имизина). Немногочисленные работы, посвященные изучению обмена катехоламинов, среди которых следует указать прежде всего на работу Стрём-Ольсена, основанную на тщательном изучении динамики обмена катехоламинов у численно небольшой труппы больных, показали, что усиленное выделение с мочой адреналина наблюдается во время маниакальных состояний, тогда как в период депрессии оно снижается — если не по сравнению с нормальными величинами, то по сравнению с маниакальной фазой. При этом разница не зависела от степени двигательной активности больных. По данным Шинфука изменения в выделении с мочой, главным образом, норадреналина, за 2—3 дня предшествовали смене фаз. В предыдущем разделе мы привели данные, свидетельствующие о большей вероятности связи стимулирующего действия ингибиторов МАО с их влиянием на обмен адреналина и норадреналина (а не серотонина). На основании преимущественно экспериментов на животных установлено и центральное адренергическое действие другого антидепрессивного средства — тофранила. Как ингибиторы МАО, так и тофранил в клинике вызывают преимущественно симптомы симпатотонии, а при передозировке нередко ведут к развитию гипоманиакальных и маниакальных состояний. Все эти данные, свидетельствуя в пользу предположения о связи изменений эмоционального состояния больных с изменением тонуса вегетативной нервной системы, вместе с тем частично противоречат гипотезе В. П. Протопопова о патогенезе маниакально-депрессивного психоза. Как известно, В. П. Протопопов полагал, что как маниакальная, так и депрессивная фаза циркулярного психоза обусловлены повышением тонуса симпатической нервной системы, что подтверждается сходством вегетативных симптомов и изменений обмена наблюдаемых в обеих фазах. Разница в клинической картине (депрессия или мания) обусловлена тем, что процесс возбуждения, возникающий в гипоталамической области, в одних случаях иррадиирует на кору головного мозга, что ведет к развитию маниакального состояния, в других — индуцирует противоположный процесс — торможение, и тогда развивается депрессия. Приведенные выше исследования подтверждают роль симпатотонии в генезе маниакальных состояний, но противоречат положению о том, что симпатотония лежит и в основе депрессии. Эффективные антидепрессивные средства — ингибиторы МАО, тофранил являются, в основном, симпатотоническими средствами. Антидепрессивное действие, хотя и менее отчетливое, обнаруживает и другая группа стимуляторов ЦНС — амфетамины или «пробуждающие» амины. Напротив, симпатолитические (аминазин и другие производные фенотиазина) и парасимпатотонические (резерпин и его производные) препараты нередко ведут к развитию депрессивных состояний. Все это говорит о том, что развитию депрессии скорее способствует снижение симпатического тонуса и что вегетативные сдвиги, участвующие в развитии мании и депрессии противоположны по своему характеру. Вместе с тем, нельзя просто отбросить данные, добытые В. П. Протопоповым и его учениками, показывающие, что при депрессивных состояниях тонус симпатической нервной системы повышен. Мы полагаем, что это противоречие может быть устранено, если предположить, что в основе развития депрессивных состояний могут лежать различные механизмы — часть депрессивных состояний связана с симпатотонией, как это и предполагал В. П. Протопопов, часть же — с парасимпатотониеи, как на это указывают результаты применения антидепрессивных средств. Хорошо известно, что эти лекарства дают терапевтический эффект лишь у части депрессивных больных, у части же состояние не изменяется или наступает обострение депрессии. Мы полагаем, что это различие в эффективности антидепрессивных средств может быть связано с различием в механизмах их возникновения. Антидепрессивные средства, повышающие тонус симпатической (эрготропной) системы должны давать хороший эффект у тех больных, у которых депрессия связана со снижением тонуса этой системы, напротив, если симпатический тонус повышен до начала лечения можно ожидать ухудшения в состоянии больных при назначении им современных антидепрессивных средств. Наш собственный опыт, пока недостаточный, говорит в пользу вероятности такого предположения. Суммируя все приведенные выше данные можно сказать, что клинический опыт оправдывает постановку вопроса о роли, которую играют адреналин и норадреналин и продукты их обмена в генезе шизофрении. Кети в своем обзоре называет эту гипотезу «заманчивой, остроумной и правдоподобной». Однако приходится согласиться с мнением, что собранные на сегодняшний день фактические данные недостаточны для того, чтобы считать эту гипотезу доказанной. Отметим, что к такому выводу приходит и Смизис, являющийся одним из создателей теории о нарушении обмена катехоламинов, как основе патогенеза шизофрении. Подавляющее большинство наблюдений, подтверждающих роль адреналина в генезе шизофрении, сделано Хоффером, Осмондом и их сотрудниками. Несомненно, более достоверными были бы данные, полученные различными исследователями, работающими независимо друг от друга. До последнего времени, однако, число подобных работ остается весьма ограниченным. Стремление Хоффера и его сотрудников, психологически вполне понятное, объяснить с помощью созданной ими теории возможно большее число явлений, наблюдаемых в клинике психических заболеваний, приводит к тому, что эта теория вызывает столь же естественный скептицизм других исследователей. Если наличие одного единственного токсического фактора, вызывающего самые различные синдромы, наблюдающиеся при шизофрении, уже представляется маловероятным, то еще менее вероятным представляется, что все тот же фактор может объяснить — пусть даже частично — возникновение хронического алкоголизма, участвовать в генезе эпилепсии, обусловливать психотомиметический эффект ДЛК. Универсализация теории делает ее менее правдоподобной и приводит к тому, что теория отбрасывается целиком, вместе с содержащимися в ней рациональными элементами. Между тем, нет и достоверных данных, позволяющих отвергнуть роль нарушений метаболизма адреналина и норадреналина в генезе психических заболеваний, в частности — шизофрении. Современное состояние наших знаний позволяет предполагать, что тонус вегетативных центров головного мозга, а следовательно и обмен одного из медиаторов вегетативной нервной системы — адреналина играет роль в развитии психических нарушений, прежде всего изменений эмоциональной сферы. Вещества, близкие по строению к адреналину и, возможно, образующиеся в организме человека, способны вызывать изменения психики, сходные с изменениями у больных шизофренией. Уточнение роли обмена катехоламинов при психических заболеваниях является задачей дальнейших исследований, которым поспешное конструирование гипотез и теорий может, наряду с некоторой пользой, принести и значительный вред. Исследования такого рода сталкиваются со значительными трудностями, т. к. данные, полученные при изучении мочи и крови больных, недостаточно достоверны, когда речь идет о процессах, происходящих на уровне центральной нервной системы. Группа экспертов Всемирной организаций здравоохранения, изучавшая этот вопрос, высказала убеждение, что адреналин (равно как и серотонин и ацетилхолин) является только представителем целой группы сходных веществ, и, может быть, представителем, не играющим решающей роли. В частности, предшественники адреналина — ДОФА, допамин могут по данным некоторых исследователей оказывать значительное влияние на ЦНС. Вполне вероятно, что в центральной нервной системе встречаются и более специфичные для нее вещества, отсутствующие на периферии. Разумеется, вряд ли можно рассчитывать, что изменения обмена только одного медиатора, будь то адреналин или серотонин, или даже нарушения их равновесия смогут объяснить всю пеструю психопатологическую симптоматику шизофрении и маниакально-депрессивного психоза, механизм действия психотомиметических средств и т. д. Однако изучение химических процессов, происходящих в головном мозгу, помогает лучше понять природу нарушений, лежащих в основе психозов, и тем самым отыскать пути их рациональной, патогенетической терапии. Создание ряда антидепрессивных средств, основанное на предположении о связи антидепрессивного действия с торможением МАО, доказывает практическую ценность такого подхода уже при современном уровне наших знаний. ЛИТЕРАТУРА1. Анохин П. К. — Физиол. ж. СССР, 1957, т. 43, стр. 1072. 2. Банщиков В. М., Столяров Г. В. — Ж. невропат. психиатр., 1963, т. 63, стр. 295. 3. Вольфовский О. И. — В кн. Лечение шизофрении, Х-в, 1939, стр. 440. 4. Гамбург А. Л. — Ж. невропат. психиатр., 1961, № 8, стр. 1216. 5. Лапин И. П., Xаунина Р. А., Щелкунов Е. Л. — Ж. невропат. психиатр., 1962, т. 62, стр. 183. 6. Утевский А. М. — В сб. Биохимия нервной системы Киев, 1954, стр. 247. 7. Утевский А. М., Осинская В. О. — Укр. біохімічн. ж., 1955, т. 27, стр. 247. 8. Шаталова А. А., Мягер В. К. — Ж. невропат. психиатр., 1960, № 10, стр. 1338. 9. Abelу P. — Ann. med. psychol., 1960, p. I, p. 1. 10. Abelу P. — Ann. med. psychol., 1961, v. 119, p. 67. 11. Abelу P., Delaville M. — Ann. med. psychol., 1960, v. 118, p. 516. 12. Abelу P., Riсhardeau N., Didieu-Anglade G., Benoit J. — CI — Ann. med. psychol., 1959, v. 1, p. 543. 13. Apter N. — Amer. J. Psychiat., 1958, v. 115, p. 55. 14. Ashbу W., Collins G., Вassett M. — J. Ment. Sci., 1960, v. 106, p. 1555. 15. Васq Z. — Pharmacol. Rev., 1949, p. II, p. 1. 16. Вeauvallet M., Fugazza J. — C. r. Soc. biol., 1960, v. 154, p. 1419. 17. Benda Ph. — Presse med., 1959, v. 67, p. 1818. 18. Вenjamin J. — Psychosom. med., 1958, v. 20, p. 427. 19. Вergsman A. — Acta psychiat. neurol. Scand., 1959, v. 34, Suppl. 133, p. 5. 20. Berthiaume M., Leduc J., D'Iorio A. — Arch. gen. Psychiat., 1960, v. 2, p. 468. 21. Blaschko H. — Pharmacol. Rev., 1954, v. 6, p. 23. 22. Brodie В., Spector S., Shore P. — Pharmacol. Review, 1959, v. 11, p. 548. 23. Вrune G., Psсheidt G. — Fed. Proc., 1961, v. 20, p. 889. 24. Carlsson A., Lindquist M., Magnusson Т., Waldeck B. — Science, 1958, v. 127, p. 471. 25. Carlsson A., Rasmussen E., Kristjansen P. — J. Neurochem., 1959, v. 4, p. 318. 26. Carlsson A., Rasmussen E., Kristjansen P. — J. Neurochem., 1959, v. 4, p. 321. 27. Cohen G., Holland В., Goldenberg M. — Arch. Neurol. Psychiat., 1958, v. 80, p. 484. 28. Cohen G., Holland В., Goldenberg M. — Arch. gen. Psychiat., 1959, v. 1, p. 228. 29. Densоn R. — Dis. Nerv. Syst., 1962, v. 23, p. 167. 30. De Schaepdryver A., Preziosi P. — Boll. soc. ital. biol. sperim., 1959, v. 35, p. 327. 31. Duner H. — Acta physiol. Scand., 1954, v. 32, p. 63. 32. EImadjian F., Hope J., Freeman H. — Arch. Neurol. Psychiat, 1957, v. 77, p. 399. 33. EImadjian F., Hope J., Lamsоn E. — J. clin. Endocrinol., 1957, v. 17, p. 608. 34. English D. — Amer. J. Psychiat., 1961, v. 117, p. 865. 35. Euler U. — Pharmacol. Rev., 1954, v. 6, p. 15. 36. Fazekas J., Thomas A., Johnsohn J., Young W. — Arch. Nieurol Psychiat., 1960, v. 2, p. 435. 37. Feldstein A. — Amer. J. Psychiat., 1959, v. 116, p. 454. 38. Fleetwood M., Diethelm O. — Amer. J. Psychiat., 1951, v. 108, p. 433. 39. Garb S., Tiwari N, Chapman L. — Amer. J. Psychiat., 1957, v. 113, p. 740. 40. Gastaldi G., De Renzi E., coll. — Rev. Pat. nerv., 1958, v. 79, p. 225. 41. Gellhorn E. — Psychiat. Res. Rep. Amer. Psychiat. Ass., 1960, v. 12, p. 209. 42. Giacobini E., Izikowitz S., Wegmann A. — Arch. gen. Psychiat., 1960, v. 3, p. 289. 43. Golse J., Morel P., Gautier H. — Ann. med. psychol., 1961, v. 119, p. 129. 44. Henneman D., Altschule M., Goncz R.-M., Davis P. — Arch. intern. Med., 1955, v. 95, p. 594. 45. Hоagland H., Bergen J., Bloch E., Elmadjava E., coll. — Jappl. Physiol., 1955, v. 8, p. 149. 46. Hoffer A. — J. clin. exper. psychopathol., 1957, v. 18, p. 27. 47. Hoffer A. — В кн. Hormones, Brain Function and Behavior, N. Y. 1957, p. 181. 48. Hoffer A. — Amer. J. Psychiat., 1958, v. 114, p. 752. 49. Hoffer A. — Dis. Nerv. Syst., 1959, v. 20, p. 597. 50. Hoffer A. — Dis. Nerv. Syst., 1960, v. 21, p. 79. 51. Hoffer A. — J. Nerv. Ment. Dis., 1960, v. 130, p. 93. 52. Hoffer A., Callbeck M. — J. Ment. Sci., 1960, v. 106, p. 138. 53. Hoffer A., Kenуоn M. — Arch. Neurol., 1957, v. 77, p. 437. 54. Hoffer A., Osmond H. — J. Nerv. Ment. Dis., 1955, v. 122, p. 448. 55. Hoffer A., Osmond H. — J. Nerv. Ment. Dis., 1959, v. 128, p. 18. 56. Hoffer A., Osmond H. — Quart. J. Stud. Ale, 1959, v. 20, p. 750. 57. Hoffer A., Osmond H. — Dis. Nerv. Syst., 1962, v. 23, p. 204. 58. Hoffer A., Osmond H., Callbeck M., Kahan I. — J. Clin. exper. Psychopathol., 1957, v. 18, p. 131. 59. Hoffer A., Osmond H., Smуthies J. — J. Ment. Sci., 1954 v. 100, p. 29. 60. Hoffer A., Рауza A. — Amer. J. Psychial.. 1960, v. 116, p. 664. 61. Hoffer A., Smith C, Chwelos N.. Callbeck M., coll. — J. clin. exper. Psychopathol., 1959, v. 20, p. 125. 62. Holland В., Соhen G., coll. — Fed. Proc., 1958, v. 17, p. 378. 63. Holtz P. — Psychiat. Neurol., 1960, Bd. 140, S. 175. 64. Holtz P., Osswald W., Stock K. — Arch. exp. Path. Pharmak, 1960, Bd. 239, p. 1. 65. King В., SokoIоff L., WeсhsIer R. — J. clin. invest., 1952, v. 31. p. 273. 66. Kopin I. — Science, 1960, v. 131, p. 1372. 67. Krupp P., Monnier M. — Experientia, 1960, v. 16, p. 206. 68. Kusсhke H., Ditfurth H. — Klin. Wschr., 1958, № 16, S. 773. 63. Lа Brosse E., Mann J., Ketу S. — Neurol. Psychiat, 1962, v. 15, p. 1432. 70. Layne D., Sоurkes T. — J. Nerv. Ment. Dis., 1960, v. 130, p. 93. 71. Lea A. — J. Ment. Sci., 1955, v. 101, p. 538. 72. Leach В., Cohen M., Heath R., Marten S. — Arch. Neurol, Psychiat., 1956, v. 76, p. 635. 73. Leach В., Heeth R. — Arch. Neurol. Psychiat., 1956, v. 76, p. 444. 74. Manger W., Sсhwarz В., coll. — Arch. Nerul. Psychiat, 1957, v. 78, p. 396. 75. McKellar P. — J. Social Psychiatry, 1957, v. 3, p. 170. 76. Mendelsоn J., Kubzansky P., coll. — Arch. gen. Psychiat, I960, v. 2, p. 147. 77. Minz В., Noel P. — C. r. Soc. biol., 1959, v. 153, p. 1937. 78. Minz В., Thuillier J. — C. r. Soc. biol., 1959, v. 153, p. 962. 79. Minz В., Walaszek E. — С r. Acad. Sci., 1957, v. 244, p. 1974. 80. Minz В., Walaszek E. — J. Nerv. Ment. Dis., 1960, v. 130, p. 420. 81. Moruzzi G. — Clin. terap., 1959, v. 17, p. 559. 82. NоvaI J., Brande В., Sоhler A. — Fed. Proc., 1959, v. 18, p. 428. 83. Osmond H. — Dis. Nerv. Syst, 1955, v. 16, p. 101. 84. Osmond H., Hoffer A. — J. Ment. Sci., 1958, v. 104, p. 302. 85. Osmond H., Hoffer A. — J. Ment. Sci., 1959, v. 105, p. 653. 86. Osmond H., Hoffer A. — Canad. M. A. J., 1959, v. 80, p. 91. 87. Osmond H., Smуthies J. — J. Ment. Sci., 1952. v. 98, p. 309. 88. Оvshinsky S. — J. Nerv. ment. Dis., 1958, v. 127. p. 180. 89. Pollin W., Goldin S. — Neurol. Psychiat., 1962, v. 15, p. 1431. 90. Quinn G., Brodie В., Shore P. — J. Pharmacol, exper. Therap., 1958. v. 122, p. 63. 91 Randrup A.. Munkvad L — Amer. J. Psychiat., 1960, v. 117, p. 153. 92. Regan P., Reilly J. — J. Nerv. Ment. Dis., 1958, v. 127, p. 12. 93. Reilly J., Regan P — Proc. Soc. Exper. Biol. Med., 1957, v. 95, p. 377. 94. Resnick O., EImadjian F. — Amer. J. Physiol., 1956, v. 187, p. 626. 95. Resnick O., Freeman H. — Arch. gen. Psychiat., 1962, v. 6, p. 388. 96. Resnick O., WoIfe J., Freeman H., Elmadjian F. — Science, 1958, v. 127, p. 1116. 97. Sakamоtо K. — Psychiat. Neurol. Jap., 1959, v. 61, p. 1497. 98. Sano I. — Biochim. Biophys. Acta, 1959, v. 32, p. 586. 99. Saunders J. — Ann. N. Y. Acad. Sci., 1959, v. 80. p. 712. 100. Shinfuku N., Omura M., Kayano M. — Yonago acta med., 1961, v. 5, p. 109. 101. Shore P., Olin J. — J. Pharmacol. exper. Therap., 1958. v. 122, p. 295. 102. Silverman A., Cohen S., Shmavonian В., Kirschner N — Rec. Advanc. Biol. Psychiat, 1961, v. 3, p 104. 103. Smythies J. — Lancet, 1958, p. II, p 308. 104. Smythies J. — Nature, 1959, v. 183, p 545. 105. Smythies J. — Lancet, 1960, p. I, p. 1287. 106. Shrom-Olsen R., WeiI-MaIherbe H. — J. ment. Sci., 1958, v. 104, p. 696. 107. Sulkowitch H., Altschule M. — Arch. Gen. Psychiat, 1959, v. 1, p. 108. 108. Sulkowitch H., Perrin G., Altschule M. — Proc. Soc. Exper. Biol. Med., 1957, v. 95, p. 245. 109. Szara S., Axelrod J., Perlin S. — Amer. J. Psychiat., 1958, v. 115, p. 162. 110. Szara S., Axelrod J. — Amer. J. Psychiat., 1960, v. 116, p. 665. 111. Szatmari A., Hoffer A., Schneider R. — Amer. J. Psychiat., 1955, v. 3, p. 603. 112. Taubmann G. — Zbl. Neur., 1956, Bd. 137, S. 142. 113. Taubmann G., Jantz H. — Nervenarzt, 1957, Jg. 28, S. 485 114. Tissоt R. — Encephale, 1961, v. 50, p. 205. 115. Udenfriend S., CreveIing С — J. neurochem., 1959, v. 4, p. 350. 116. Vander W., SpoerIein M. — Arch. internat. pharmacodyn., 1962, v. 137, p. 145. 117. Veech R., Altschule M., Sulkowitch H., Holliday Ph. — Arch. gen. Psychiat., 1960, v. 3, p. 642. 118. Veech R., Вigelow L., Dencula W., Altschule M. — Arch. gen. Psychiat, 1961, v. 5, p. 127. 119. Vоgt M. — Pharmacol. Rev., 1954, v. 6, p. 31. 120. Vojtechovsky M., Grof S., Vitek V. — Csl. psychiatr., 1962, v. 58, p. 383. 121. Weil-Malherbe H. — J. Ment. Sci., 1955, v. 101, p. 733. 122. Weil-Malherbe H., Bone A. — J. Endocrinol., 1954, v. 11, p. 285. 123. Weil-Malherbe H., Bone A. — J. Endocrinol., 1954, v. 11, p. 298. 124. WHO — Technical report series № 152, Geneva, 1958. 125. Yura R. — Zbl. Neurol., 1961, Bd. 161, S. 255. Примечания:5 Метод аутостимуляции заключается в том, что животные с электродами, подведенными к определенному участку головного мозга, сами нажатием на рычаг могут либо замыкать ток (большая частота замыканий расценивается как показатель «приятного» характера раздражения), либо прерывать неприятные импульсы. При аутоинъекции животных таким же образом регулируют повторные поступления в мозг малых количеств исследуемого вещества. 6 Антиметаболит по Вулли — вещество, обнаруживающее структуру, сходную со структурой метаболита, соединяющееся с теми же рецепторами, но не оказывающее специфического для метаболита действия. В результате развивается состояние, характерное для недостатка данного метаболита. Вулли сравнивает метаболит с ключом, отпирающим дверь, а антиметаболит — с ключом, который входит в замочную скважину, но дверь не отпирает. |
|
||
|
Главная | В избранное | Наш E-MAIL | Прислать материал | Нашёл ошибку | Верх |
||||
|
|
||||